|
THE PHYSICS
AND PATHOLOGY OF WOUNDS PART 2: AUTO-IMMUNOPATHY AND THE INTRINSIC DISEASE
OF WOUND HEALING Marc E. Gottlieb, MD, FACS Phoenix, AZ Revision
01a, February 10, 2010, Copyright © 2010 |
|
|
Preamble In Part 1 of this series, The Wound as a System and a
Controlled Machine, the main point was that the wound is not only a complex
system, but it is a non-linear controlled system. Control is the basis for all wound
dynamics, allowing the healthy wound to heal by one-shot dynamics, but
leading to complex patterns when wound healing is pathological. Here in Part 2, Auto-Immunopathy and
the Intrinsic Disease of Wound Healing, we will now go from a
physics-engineering perspective to a clinical-pathological one. Here we will show that the wound, made of
fibroblasts and angiocytes, is just an instance of the general fibrous stroma
(trying to repair itself after injury).
As such, the wound is subject to the same diseases as the general
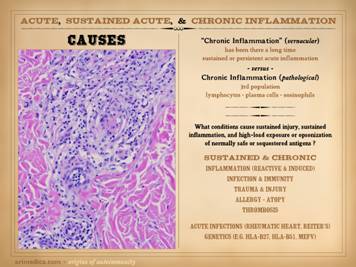
stroma, which mainly means the auto-immune connective tissue disorders. These conventional diseases and the chronic
wound can be equated through the principle of sustained chronic inflammation
leading to immune sensitization against stromal elements. In the next part, Part 3, Chronicity
and the Intrinsic Disease of Wound Healing, we will bring together the
engineering aspects of the wound as a controlled process and the
clinico-pathological aspects of stromal auto-immunization to understand why
chronic wounds fail to heal, why the wound control loop cannot succeed in the
face of intrinsic auto-immune wound chronicity. |
|
|
|
|
|
In Part 1, we looked at the wound as a system subject to
feedback and control. This model of
the wound can accommodate all conditions of wound physiology and pathology,
of normality, failure, and therapy.
Part 2 will now look at more conventional biological and medical aspects
of wound. The focus will be on
intrinsic wound pathology and failure, specifically the condition of
intrinsic degradation of wound healing not attributable to extrinsic factors. This intrinsic disease of wound healing
results from the appearance of an abnormal population of chronic inflammatory
and immune cells which has complex disruptive effects on the two cell sets
which belong there, acute inflammation and wound module. These states have a critical cause-and-effect
association with autoimmunity, microthrombosis, and other events which
sustain acute inflammation. The
auto-immune connective tissue disorders are one of the common causes of
chronic ulceration, ulcers and wounds which can be especially refractory to
treatment. We will see here that
connective tissue disorders and the chronic intrinsically pathological wound
are essentially the same thing, auto-immune attack directed against the
fibro-vascular stroma made by fibroblasts and angiocytes. |
|
|
|
|
|
This is an editorial on one of the core competencies of wounds
or any other practice in medicine – the necessity of and ability to make a
proper diagnosis. All good care starts
with a correct diagnosis. This is one
of the supreme obligations of all physicians, especially specialists who are
expected to have greater knowledge of specific subjects. This includes wounds and wound
practice. Here are some basic
principles of medical practice as relates to the full spectrum of chronic
wounds. (1) Chronic and pathological wounds (CAP wounds) are a
distinctive class of disease and clinical activity. (2)
There is a non-expert legacy misunderstanding of wounds that focuses only on
trauma and a few standard categories of CAP wounds - arterial, venous,
pressure, diabetes. (3) Legitimate purveyors, professors,
and practitioners of this specialty must have the professional level of
knowledge required to master these diseases.
Like all specialties, this starts with an understanding of the full
spectrum of relevant pathologies. (4) What are often dismissed as
"atypical wounds" are not atypical at all. In fact, they are the core of chronic and
pathological wounds, and they are far more abundant and significant than
naives and non-experts perceive. |
|
|
|
|
|
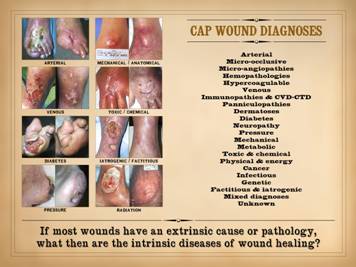
CAP wounds: Chronic and
Pathological Wounds. This is the
subject of modern wound care. These
are not the simple injuries for which normal physiological wound healing
turns on and resolves the problem.
These are not the simple trauma and surgical wounds in healthy people
that will heal anyway. These are not
the wounds that heal by simple convergent one-shot dynamics. These are the sick wounds that cannot heal. These are the wounds due to pathology and
disease. CAP wounds result from 2
general problems: (1) wounds that are caused and maintained by some sort of chronic
illness or pathology; (2) wounds, from whatever primary cause,
that fail (fail to heal, are wound healing incompetent) due to disease or
disorder of the wound healing process. |
|
|
|
|
|
There are many primary diseases or diagnoses which lead to
chronic, pathological, non-healing wounds and wound complications. Major diagnostic categories, concepts, and
groups include: arterial,
micro-occlusive, micro-angiopathies, hemopathologies, hypercoagulable,
venous, immunopathies & collagen vascular / connective tissue disorders
(cvd-ctd), panniculopathies, dermatoses, diabetes, neuropathy, pressure,
mechanical, metabolic, toxic & chemical, physical & energy, cancer,
infectious, genetic, factitious & iatrogenic, mixed diagnoses,
unknown. These various diseases cause
the wounds or else inhibit the wounds from healing. One of the fundamental principles of wound
pathology and practice is understanding the difference between (1) a
sustained or perpetuated wound due to some extrinsic applied disease or
injury versus (2) an intrinsically disordered wound in which the
machinery of repair is itself damaged or pathological. Consider arterial disease.
It affects wound healing, but it is not a disease of wound
healing. Patients with
athero-occlusive ischemia of the feet can heal wounds just fine anywhere
else, and they heal their foot wounds as soon as revascularization restores
blood flow. Pressure ulcers are
trauma, and such patients heal any wound other than those subject to the
repetitive injury of prolonged positional ischemia. Neuropathic wounds occur because of altered
skeletal biomechanics leading to pressure without the protective sensation
and mobility needed to avoid injury. Diabetic
ulcers are a multifactorial mix of arterial, pressure, neuropathic, and
biomechanical factors, and if you do a thyroidectomy or fix a broken wrist,
things heal properly because the risks for diabetic ulceration are not global
affects on the machinery of wound healing.
Radiation ulcers are notoriously problematic, because radiation does
kill the machinery of wound healing, but this is a local trauma, not a
system-wide deficiency of wound healing biology. Do you see the trend here?
The chronic wound examples just cited are attributable to some disease
or injury extrinsic to the innate wound healing system. It all begs the question: What then are the intrinsic diseases of
wound healing? Given that every other
cell, tissue, organ, and system in the body is subject to some greater or
lesser affliction, why then do we not recognize or acknowledge those diseases
of the wound healing system? Surely
they must exist. The main purpose of
this whole presentation is precisely that, to illuminate these diseases for
you. This is crucial to proper care,
because choosing effective therapies is contingent on correct diagnosis. True intrinsic diseases of wound healing
exist. They are common. Unfortunately, they are naively overlooked
and pigeon-holed into the few diagnostic categories that non-experts are
aware of, such as arterial and venous.
Understanding intrinsic wound disease begins by understanding the
basic anatomy and function of the wound: a self-reorganization of
fibroblasts, angiocytes, and epithelium.
We looked at the basic biology of this process in Part 1. Here, we will look at how this
fibro-vascular system becomes altered and pathological. |
|
|
|
|
|
To understand how wound healing becomes sick, you must first
understand what wound healing is. In
Part 1 we looked at the tangible physical details of what happens in the
healing wound, the cells and chemicals, and we looked at their operational
dynamics. But there is something more
rudimentary and central to the wound, the “heart and soul” of the process. Ask these two questions: (1) What is the quintessential
structure & function of the wound?
(2) What is the quintessence of its dysfunction? To answer these, first think about other
more obvious organs. Whatever its
complex anatomy and physiology, at its core the heart is a pump. When it fails, it is an inadequate pump
that dams the circulation. The lung is
a bellows and diffusion membrane. When
it fails, there is inadequate movement of gases causing impaired ventilation
and respiration. The kidney is a
filter and resorption membrane. The
eye is a light collector and detector, the ear a sound transducer and decoder. The skeleton is a set of structural members
and linkages, the nerves an electrical control network. When these organs fail, they fail to serve
their quintessential functions. What is the quintessential structure & function of the
wound? It is not a pump. It is not a diffusion surface. It is not a structural support framework,
nor a sound or light collector, nor a command and control center. It is not an endocrine regulatory agent,
not a secretory system for protection or digestion, and not a set of conduits
for bulk transport. The wound is an
evanescent structure that comes when needed, then disappears. It is nothing more than a set of cells
whose task it is to restore continuity of the tissues whenever there has been
an injury – a re-epithelialized
parenchymal matrix. What then is the
wound? It is a self-reorganizing set
of cells. What is the quintessence of its
dysfunction ? It fails to reorganize –
a failure of logistics and inter-operation. This subject will be addressed in greater detail in Part 3,
where the focus will be on how the wound fails. For now though, here in Part 2, as we look
at conventional clinical wound healing diseases, keep the following points in
mind. The wound module is a special ad
hoc reserve organ. What are the
quintessential structures and functions of the wound? It is a set of mutually interactive cells
that self-organizes to recreate generic stroma. It has no function other than organizing
itself into the generic stroma that is the foundation for other tissues and
organs. What is the quintessential
derangement of intrinsic wound pathology and chronicity? It is a disorder of “logistical”
self-re-organization, failing to restore the intended stroma. When it fails, it simply fails to assemble
into its intended final form, to complete its task to become something and
then cease. |
|
|
|
|
|
Understanding conventional pathology and clinical disease begins
with a firm understanding of normal anatomy and physiology. Thus, here is a reminder about wound
healing physiology, as reviewed in Part 1.
The “wound” as discussed here is not the injury or defect, but rather
the post-injury post-inflammatory wound module of proliferative repair, that
ad hoc organ that arises to repair the damage and restore a closed
(epithelialized) generic stroma. Right: a healthy wound goes through the natural
process of healing until closed, i.e. epithelialized. Left: all of the reparative events taking place
in the wound have a well organized and recognizable anatomy. Each of the histologic strata correlates
with the phases of repair and grossly observable events. Seven distinctive findings can be observed
clinically that attest to this process: 0 – Injury and the
body’s defenses against it, thrombosis and inflammation, are the triggers
which initiate the repair process.
Sustained injury creates conditions which suppress repair. Injury, thrombosis, and inflammation must
be alleviated if the wound module is to develop. 1 – Inflammation
subsides. This is the indication that
injury has abated and that the defensive response to injury is subsiding,
permitting the onset of repair events.
Microscopically, inflammation is still present – it is what drives the
control loop of repair – but clinically the acute phase intense defensive
events with manifestations of erythema, edema, pain, warmth, drainage, etc.
will subside before or as repair turns on. 2 – Macrophages appear
in the wound, observable clinically by the separation of eschar. Their efferent function, to make
proliferative cytokines and drive the system load, the repair cells, becomes
manifest when “granulation tissue” and other signs of active healing appear. 3 – Aminoglycan
ground substance appears, as the early medium required by the early repair
cells, the space in which they will make a structural stroma. It is evidenced clinically by mucus on or
just under the surface of the wound. 4 – “Granulation
tissue” is the conventional name used to describe the appearance of
angiogenic new blood vessels within the new aminoglycan medium. The saturated red color seen clinically is
due to a high supra-physiological vascular density (for the reasons discussed
in detail in Part 1). Angiogenesis is
required to restore an environment and supply network before fibroblasts can
start to produce a fibrous matrix. 5 – Histioblasts
and fibroblasts are the cells that come in behind the angiocytes to make the
fibrous proteins and matrix that gives structural competence to the new
stroma. Fibroplasia is evident
clinically by stiffness, loss of compliance, and increasing strength in the
wound. 6 – Myofibroblasts
are the specialized fibroblasts that have accumulated contractile muscle
proteins, allowing them to contract the developing matrix, thereby reducing
the size of the wound and defect.
Their effects are evident clinically as reduction in area of the
wound, and deformations of the wound margins. 7 –
Epithelialization is the final component of the active phase of repair. This is the tangential growth of the
epithelium (or other ecto-entodermal parenchyma) over the new mesenchymal
stroma. Once the epithelium is
completely confluent and continuous, the wound is “closed”, the nominal
clinical endpoint of healing. 8 – Maturation. Once the wound is epithelialized and
closed, the control loop stops driving and acute phase wound healing
ceases. The supra-physiological
accumulation of vessels and matrix within the new stroma – the scar – now
begins a slow process of teardown and remodeling until the new stroma
eventually resembles normal dermis or fascias. |
|
|
|
|
|
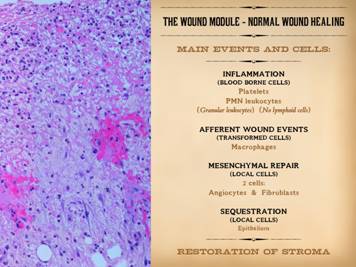
This slide is to remind that the integrated response to injury –
inflammation then wound healing – has a few main events and agents. Injury is recognized, and the inflammatory
response is activated, by blood borne cells, the platelets and granular
leukocytes. Aside from its various
host defense and cleanup functions, inflammation also initiates wound healing. This occurs via the transformation of blood
borne monocytes into their tissue active phenotype, the macrophage. Macrophages regulate the afferent wound
events, the activation and attraction of local mesenchymal cells, the
angiocytes and fibroblasts. These cells
in turn are the effectors of the efferent wound events, the creation of the
new stroma composed of blood vessels and connective protein matrix. Sequestration of the mesenchyme from the
ambient world by growth and confluence of the overlying epithelium marks the
nominal endpoint of wound healing. The
purpose of all of this is to restore and re-epithelialize a generic stroma. However, this post-inflammatory wound
healing version of the stroma is “scar”, excessively dense in the products of
repair. Maturation is the post-healing
phase of the process in which the excesses are remodeled back to a normal dermal
or fascial form of stroma. |
|
|
|
|
|
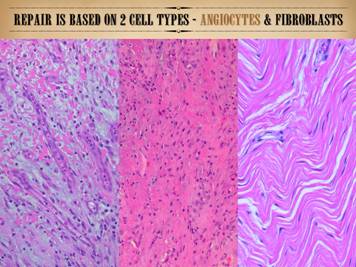
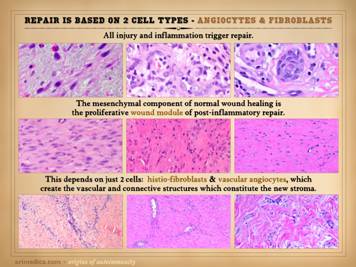
Not to be redundant or repetitive, but it is important to
remember that the mesenchymal component of scar is based on two cell types –
angiocytes which make new vessels, and fibroblasts which make new connective
matrix. Angiocytes and fibroblasts,
blood vessels and connective matrix – that is the generic stroma (which
includes dermis, fascias, miscellaneous connective tissues, and the wound). Left:
in the angio-organization layer, angiocytes have formed long vertical
cords and conducting vessels, reaching from mature vessels underneath toward
chemotactic stimuli above. Once these
vessels and blood flow are established, the fibrous component of repair can
start to develop. Center: numerous fibroblasts are making fibrous
collagen, giving the new stroma mechanical stability. The stratification,
condensed organization, and dense packing of the collagen fibers is obvious. Right: a matured scar (years old) with open
compliant collagen bundles and sparse vessels and fibrocytes, much closer to
normal dermis or musculotendinous fascias rather than scar. Regardless of the phenotype, subtype,
“breed”, or “avatar” of the stroma – dermis, fascia, wound, synovium, etc, –
the elemental constructs are the same:
angiocytes, fibroblasts, vessels, connectives. |
|
|
|
|
|
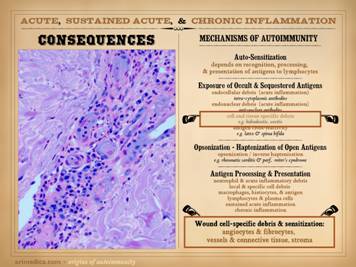
We have just emphasized the central role of angiocytes and
fibroblasts to make the generic stroma of the body, both during embryonic
development, and also during wound healing, which is simply the restitution
of the stroma after injury. Consider
for a moment what diseases might affect this stroma, either the angiocytes
and fibroblasts which make it, or the vessels and connective matrix which are
the result. Are you aware of any
common metabolic diseases of these cells and structures? Sure, there are interesting problems of
collagen, such as Ehlers-Danlos syndrome, but that is a collagen problem per
se, not a fibroblast problem, and either way, it is an oddball diagnosis in
the big picture of things. Arterial
atherosclerosis is a common problem, but it is a complex degenerative process
of the composite structure of large blood vessels, not a disease of
individual angiocytes and the smaller vessels they make. But are there metabolic or genetic or
degenerative diseases of fibroblasts and angiocytes that make them fail to
function correctly? Even in wounds
which are highly impaired, the host can still heal wounds without impediment elsewhere
on the body, for example a patient with a chronic ankle ulcer will heal just
fine after a humerus fracture or thyroidectomy or cholecystectomy. As will be discussed in detail in
subsequent slides, the answer simply is “no”, there are no common diseases
intrinsic to these stromal cells. What
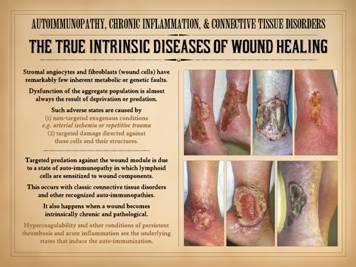
does affect them adversely are conditions of exogenous deprivation or predation. Those adverse conditions which would deprive or attack the
stroma can be (1) non-targeted (not explicitly directed against stromal
elements) exogenous conditions, e.g. arterial ischemia or repetitive trauma,
and (2) targeted damage directed against these cells and their structures. What can pointedly attack the connective
stromas and their constituent cells?
Their names say it all – the “collagen-vascular diseases” and the
“connective tissue disorders”. Targeted
predation against the stroma, and hence against the wound module is due to a
state of auto-immunopathy in which lymphoid cells are sensitized to wound components.
This occurs with classic connective tissue disorders and other recognized
auto-immunopathies. As will be
presented here, anti-stromal autoimmune sensitization also happens when a
wound becomes intrinsically chronic and pathological. Hypercoagulability and other conditions of
persistent thrombosis and acute inflammation are the underlying states that
induce the auto-immunization. |
|
|
|
|
|
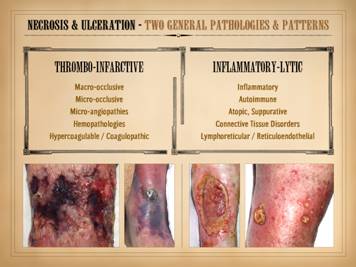
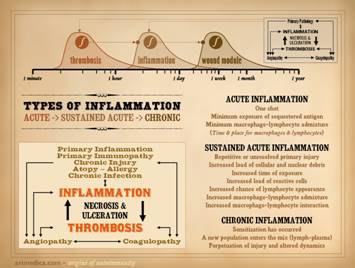
“Necrosis” and “ulceration” are the two main words that describe
the active onset and evolution of a pathological wound. In trying to understand the intrinsic
diseases of healing and the persistence of CAP wounds, the first major
concept to understand is that there are two patterns and final pathways to
necrosis and ulceration – the thrombo-infarctive pattern and the
inflammatory-lytic pattern. (1)
Thrombo-infarctive necrosis and ulceration is a consequence of severe
ischemia due to loss of blood flow.
Obstructed circulation of any cause can be responsible. Large vessel macro-thrombosis with large
organ infarcts or limb gangrene is one version of this which clinically is
usually overt and obvious. With regard
to non-life threatening pathological ulceration and CAP wounds, the primary
problem is usually obstruction of micro-vessels, causing small scale ischemia
and infarction. This results from the
various micro-occlusive disorders, including micro-angiopathies, formed
element hematopathologies, and hypercoagulopathies and dysproteinemias. Clinically, the pattern is one of dry
gangrenous infarction, including dry eschar, cyanotic vascular stasis or else
pallor, and absence of edema and gross inflammatory changes. Laboratory measures of perfusion, such as
TcpO2, laser doppler, and multi-spectral imaging are likely to show
impairments. The underlying cause might
not always be obvious, the patient might not have an overt or established
history of a causative disorder, and the clinical presentation might not be
dramatic or life and limb threatening.
However, the physical findings are distinctive, which is enough to
direct the clinician to focus on the occlusive disorders as main items on the
differential diagnosis. (2)
Inflammatory-lytic necrosis and ulceration is due to active inflammatory
states, including primary neutrophilic inflammation, atopic-allergic inflammation,
and immune-lymphocytic inflammation, all resulting from various underlying
diseases including the autoimmunopathies, collagen-vascular connective tissue
disorders, and lymphoreticular diseases.
Immunoglobulins, complement, and matrix proteases are abundant along
with other acute inflammatory chemistry.
Clinically, these are ulcers which have overt acute inflammation,
including edema and scarlet red erythema.
Rather than having dry infarcts and eschar, these ulcers simply erode,
getting larger by the literal dissolution of the tissue by cell killing, complement-antibody
and other cytolytic events, and protease and other destructive effects. Many CAP ulcers are obviously of one origin
or the other, predominantly thrombo-occlusive versus inflammatory-lytic, and
thus they can be easily discriminated by simple physical exam as to which
underlying pathology predominates.
However, because of the intimate and intricate inter-dependence of
inflammation and thrombosis, many ulcers will have features of both patterns. There is a third major pattern of ulceration, trauma, which
includes simple mechanical or surgical injury along with pressure, radiation,
burns, toxic chemicals, etc. What
discriminates trauma as a cause of a wound is that trauma is incidental and
self-limited, whereas thrombo-infarctive and inflammatory-lytic ulceration
are generally persistent and long-lasting due to active ongoing disease (and
mutual sustentation when both patterns and pathologies are present). As will be explained further in later
slides, angiocytes and fibroblasts, the two constituent cells of the generic
stroma and wound healing process, are robust, with extraordinarily few
intrinsic diseases and pathologies.
They can be obliterated by trauma, by critical deprivation of blood
supply, and by killers such as antibodies, leukocytes, and lymphocytes. Aside from the trauma causes of wounds,
thrombo-infarctive and inflammatory-lytic ulceration and necrosis are the two
– and the only two – common pathophysiological mechanisms by which the basic
stroma of the body can be killed and degenerated. |
|
|
|
|
|
Reviews of
essential subjects - Hypercoagulopathy As a preamble to understanding CAP wounds and the intrinsic
disease of wound healing, three relevant subjects must be reviewed: the hypercoagulable disorders and ulcers,
the autoimmune connective tissue diseases and ulcers, and the basic anatomy
and cellular biology of wound healing.
The basic anatomy and biology of wound healing was presented in Part
1, and was reviewed in the last few slides.
The next set of slides will review hypercoagulability, the
hypercoagulable disorders, and the necrosis, ulceration, and CAP wounds that
result. Hypercoagulability is one of the major categories of chronic and
pathological ulceration. This subject
started to appear in published journals in the early to mid 1990’s, so after
15-20 years it can hardly be considered new.
However, it is still arcane in the sense that most practitioners
remain largely unaware of it. So, to
reiterate, hypercoagulable wounds are a MAJOR category of CAP wounds. As will be shown later, these are one of
the major groups of primary disorders which can then lead to secondary
auto-immune wound failure. This short
introduction will cover just the essentials.
Much more information on this subject can be found on the website arimedica.com, under the category
“Coagulopathies”. |
|
|
|
|
|
The next few slides are a brief introduction to
hypercoagulopathy and the related thrombotic and micro-occlusive disorders
that they cause. Recall what the
function of thrombosis is – to stop bleeding from injured blood vessels. The plasma protein blood coagulation
system, along with platelets, is tuned so that ideally the system is never
activated when flowing blood is looking at normal endothelium, but it triggers
and auto-amplifies quickly when the system “sees” any extra-vascular
chemistry or histo-anatomy. All
physicians have some familiarity with what happens when the system is untuned
toward a hypocoagulable state, with hemorrhagic risks or events resulting
from trauma, hemorrhage, sepsis, factor deficiencies, marrow suppression,
anticoagulant drug effects, etc.
Hypercoagulability remains largely misunderstood, unknown, or under
appreciated, even though it is common and has a variety of significant
clinical syndromes and sequelae.
Hypercoagulable states have a wide spectrum of etiologies which can be
primary (e.g. gene mutations) and secondary, including induced (e.g.
auto-immune thrombogens) and reactive (e.g. anti-thrombin proteins). They also have a wide spectrum of clinical
sequelae, syndromes, and presentations. The quintessential fault in hypercoagulable states is that the
blood coagulation system is over-tuned, likely to trigger and clot with
normally sub-threshold stimuli, including spontaneous thrombosis within uninjured
blood vessels, and overly aggressive thrombosis following injury. The clinical consequences may involve large
vessels and organs and be acute and overt.
They may involve small vessels and be subtle, occult, persistent, and
hard to recognize or treat.
Hypercoagulable states can be grouped among a broader category of
disorders, the thrombo-occlusive and micro-occlusive disorders. These are a consequence of pathology or
alterations in hemodynamics, blood vessels, the various other components of
whole blood, and the plasma protein clotting system itself. Various combinations of these causes can
occur, and vascular and hematological disorders superimposed on a background
hypercoagulable state can be especially problematic. While the large vessel complications of
hypercoagulability are themselves a major subject, with regard to chronic and
pathological wounds our interest is in micro-thrombosis and
micro-occlusion. The micro-thrombotic
disorders can be grouped by a few major pathophysiological mechanisms: 1 – Hemodynamic
disorders: Blood vessels, blood,
and coagulation are all intrinsically normal.
Thrombosis occurs from blood stasis due to hemodynamic alterations
related to gross cardiovascular anatomy and function (e.g. atrial
fibrillation, valvular pathology, vascular compression). 2 –
Endovasculopathies: Intrinsic and luminal
vasculopathies in which blood vessels are abnormal. Blood is normal, and coagulation is
intrinsically normal. Thrombosis occurs
in response to blood stasis or thrombotic activation created by endoluminal
and endothelial alterations in the vessels (e.g. atherosclerosis,
hyperparathyroidism-calciphylaxis). 3 –
Exovasculopathies: Extrinsic and mural
vasculopathies in which blood vessels are abnormal. Blood is normal, and coagulation is
intrinsically normal. Unlike the
endovasculopathies in which thrombosis is triggered by thrombogenic surfaces
and flow turbulence or stasis, the exovasculopathies tend to be inflammatory
or immune in origin, with inflammatory mediators triggering thrombosis in
passing blood (e.g. venous vasculitis, the connective tissue disorders, and
classic arteritides such as polyarteritis nodosa and thromboangiitis
obliterans). 4 –
Non-hypercoagulable hemopathologies:
Micro-occlusive disorders in which vessels are normal and the plasma
protein coagulation system is intrinsically normal, but other elements of the
blood are abnormal. The clotting
system responds “correctly” to abnormal conditions of stasis or thrombotic
activation (1 - hemoglobinopathies, e.g. sickle, thalassemia, hemolytic
anemias; 2 - dys- and
cryoproteinemias, e.g. cryoglobulins, cryofibrinogen, macroglobulins,
gammopathies & myeloma; 3 - red
cell, leukocyte, & platelet abnormalities, e.g. spherocytosis,
myeloproliferative disorders, polycythemias, leukemias). 5 –
Hypercoagulable hemopathologies:
Vessels are normal. Blood is
normal (formed elements and serum).
What is abnormal is the plasma protein clotting system. In the above categories, the clotting
system is behaving properly in response to abnormal conditions. In the hypercoagulopathies, abnormal
inappropriate thrombosis is the primary event. Blood stasis and vascular occlusion are
consequences, not causes. The
hypercoagulable disorders can be intrinsic (the “pre-thrombotic” primary
disorders of the coagulation system) or extrinsic due to metabolic or
auto-immune alterations. See the
following slides for specifics. The hypercoagulable states can cause both large vessel thrombosis
and micro-thrombosis. “Old medicine
syndromes” due to macro-thrombosis, such a coronary or cerebrovascular
occlusion, femoro-popliteal embolism, pulmonary embolism, and Budd-Chiari
hepatic thrombosis are overt, dramatic, and easy to recognize. Micro-thrombosis tends to be subtle,
ongoing, frustrating, and easy to overlook, misinterpret, or
misdiagnosis. One point worth
remembering is the clinical syndrome of occult hypercoagulopathy. It is a dependable tetrad or pentad of
features, and if on history alone your wound patient has these things (not
all of them need to be present), then they have a hypercoagulable
disorder: 1 – history of thrombotic or embolic
events; 2 – history of
miscarriages; 3 – history of
wound pathergy (unexpected wound complications following trauma or surgery)
or soft tissue problems including chronic ulceration; 4 – an auto-immune or connective
tissue disorder; & 5 –
(what makes it the pentad) a family history of the main 4 counts equally as a
positive personal history. |
|
|
|
|
|
This slide lists relevant basic pathological features of the
hypercoagulopathies. These lists are
not comprehensive. The hypercoagulable
disorders can be intrinsic (“pre-thrombotic”) primary disorders of the
coagulation system or extrinsic due to metabolic and auto-immune
alterations. They have important
associations with other diseases and clinical syndromes. They can cause commonly recognized large
vessel thrombotic and embolic events, or poorly recognized micro-thrombotic
events. Prethrombotic disorders, related disorders, & disease
associations: Common intrinsic causes
are gene mutations (e.g., factor 5 Leiden, prothrombin 20210G), coagulation
protein alterations (e.g., proteins C & S, anti-thrombin-3, plasminogen,
fibrinogen), and various other pathologies with a tie-in to the formation and
metabolism of these factors (e.g. liver disease, estrogens and pregnancy,
paroxysmal nocturnal hemoglobinuria, dicoumarol-derivative
complications). The extrinsic causes
include miscellaneous metabolic and pathological states (e.g. homocysteinemia
and cancer-Trousseau), but they are dominated by the antiphospholipid
antibody syndromes and other immune thrombogens and auto-immune states
(including the anticardiolipins and the lupus anticoagulants). Virtually all of the classic connective
tissue or collagen-vascular diseases have a high incidence of
hypercoagulopathy, and vice versa. The
importance of the gene mutations must be emphasized. You cannot cheat on a gene test, so when a
patient has an altered gene and then a bunch of other syndromic clinical
problems, it is a good bet that the genetic mutation is the root cause. These last two points, concerning
prethrombotic gene mutations and the connective tissue disorders will be
discussed in much greater detail in later slides. To emphasize how this knowledge must change traditional
practices, consider venous disease.
The hemodynamics of venous reflux and hypertension have been
understood for well over 200 years, yet altered hemodynamics alone do not
explain the whole picture of chronic venous ulceration. Why do these people get thrombosis and
damaged valves in the first place? Why
are their wounds hard to heal? The
answer is that many of them have factor V Leiden, prothrombin 20210G, or
another of the hypercoagulable entities as the primary underlying cause. The hypercoagulable disorder causes the
overt or occult venous thrombi that cause valvular incompetence and the
post-phlebitic state, and then when liposclerosis and ulceration eventually
ensue, the hypercoagulability also impairs healing. Macrothrombosis:
Hypercoagulable states can cause both large vessel and small vessel
thrombosis. Large vessel thrombotic
events cause the major infarcts and apoplexies that present as easily
recognizable clinical syndromes (e.g. cava-tibial venous thrombosis,
aorto-tibial arterial thrombosis, other peripheral thrombosis, coronary
artery thrombosis, cerebrovascular thrombosis, pulmonary embolism,
intracardiac thrombosis, graft and valve thrombosis, subclavian vein (paget-schroeder),
hepatic veins (budd-chiari), pituitary apoplexy (sheehan), retinal artery
& vein occlusion, intracranial sinus thrombosis, spinal apoplexy,
visceral apoplexy (renal, adrenal, bowel), etc.) These items on the list of large vessel
vascular events have one thing in common – they are acute, overt, and
life-and-limb threatening. Microthrombosis:
In comparison, microthrombotic events are subacute and chronic, slow,
subtle, persistent, recurring, perplexing, frustrating, refractory. They are non-obvious in origin – vascular
occlusion not overt – unless you are familiar with their spectrum of disease
and secondary clinical events. Some
clinical events ought to raise immediate “red flags” of a possible underlying
hypercoagulable disorder, e.g. non-immune glomerulonephritis, primary
pulmonary thrombosis, warfarin necrosis, complications of contraceptives, and
(of course) certain non-healing ulcers and thrombo-infarctive complications
of surgery. Other miscellaneous
features which raise suspicion include refractoriness to treatment or a long
history of failed treatment, any age, and family history. Remember the diagnostic, nearly
pathognomonic tetrad: 1 – thrombosis or embolism; 2 –
miscarriage; 3 – wound pathergy (including chronic ulcers); 4
– auto-immune disorder; (& 5 – personal or family history). |
|
|
|
|
|
Like anything else in medicine, proper diagnosis of a wound or
underlying disease starts with a history and physical exam, formulation of a
differential diagnosis if the exact diagnosis is not yet evident, then
resolution of the diagnosis by further testing. History was covered on the preceding
slide. This slide concerns what is apt
to be found on examination, both the initial physical plus subsequent
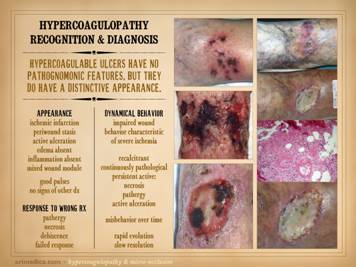
observations as treatment is managed. Hypercoagulable ulcers have features predominantly attributable
to ischemia and arterial insufficiency.
To the extent that they might have an associated immune component,
there may be inflammatory changes along with ischemic changes. However, for prototypical coagulopathic
ulceration, the pattern is one of thrombo-infarction rather than
inflammation-lysis. This means that
they have no unique nor pathognomonic features, but they do have an eminently
distinctive appearance. Features of gross appearance include: ischemic infarction (black desiccated
eschar), periwound vascular stasis (cyanotic plethora as opposed to the
scarlet hyperemia of inflammation), active ulceration (observable at the
margins where skin is dying, until the cause of ischemia has been corrected),
absence of edema, absence of gross inflammation, and a weak or absent wound
module. Unlike with classic arterial
diseases, patients will have these signs of arterial ischemia while still
having good pulses. If a patient has a
related condition, such as secondary venous disease caused by the chronic
hypercoagulopathy, then exam can be mixed with signs of the multiple
problems. However, for paradigm
hypercoagulable ulceration, the picture is one of localized arterial ischemia
in the face of good pulses. Observations over time and care, until definitive treatment is given,
can be summed up simply as “impaired wound behavior characteristic of severe
ischemia “. Wound behavior is
continuously pathological, with persistent active necrosis, pathergy, and
active ulceration. Wounds are
recalcitrant, with impaired dynamics and failure to make meaningful progress
until ischemic conditions are relieved.
Repetitive occult micro-thrombotic events result in rapid evolution
and slow resolution of the ulcers. If
wrong therapies are attempted based on wrong diagnosis, if no precautions are
taken to prevent or mitigate thrombosis and ischemia, then no results or
contrary results will happen. This is
especially problematic for attempted surgery which will fail due to
thrombo-infarctive wound and soft tissue complications such as necrosis and
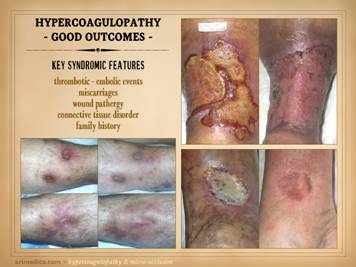
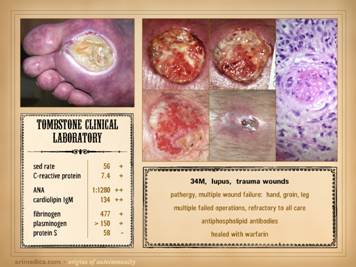
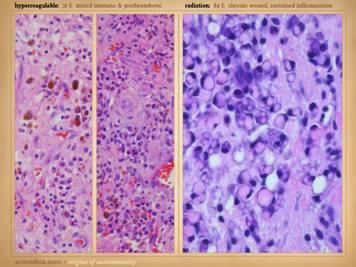
dehiscence, i.e. wound pathergy. Left upper: multifocal ankle infarcts in a patient with
protein C and anticardiolipin abnormalities.
Note the black eschar, absence of lytic ulceration and tissue
dissolution, and absence of generalized edema and panniculitis beyond the
immediate zone of the skin infarcts. Left center: distal leg ulcers in a patient with good
ankle pulses and anti-thrombin-3 deficiency.
Note dry black skin infarcts and eschar, vascular stasis and cyanosis,
absence of edema, in fact with wrinkles due to desiccation, all consistent
with severe micro-occlusive ischemia. Left lower: wound infarcts with acute black eschar, in
a forearm wound, in a patient with rheumatoid and proteins C & S
abnormalities. Right: ulceration of the
ankle after biopsy of a small lesion, in a patient with protein C deficiency
and positive cryoglobulins. Note
absence of generalized edema and inflammation, a caput medusa or venous
“spider” consistent with prior thrombosis and valvular reflux, and the
histologic findings of thrombosis and vascular necrosis. |
|
|
|
|
|
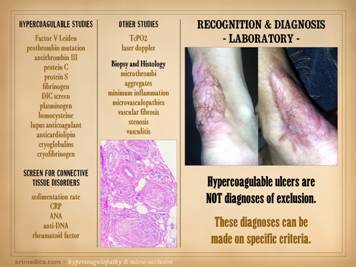
Once a diagnosis of a hypercoagulable ulcer or hypercoagulable
state is suspected, the diagnosis can be refined or confirmed by laboratory
evaluation. There is a caveat here
though. We only have clinical lab tests
for perhaps a dozen or two chemical species involved in this problem, whereas
the problem can involve many dozens or hundreds of items. A positive diagnosis is not contingent on a
positive laboratory test. This is akin
to the evaluation of connective tissue disorders. Many such patients are sero-negative. If a patient comes in with crippling wrist,
hand, ankle, knee, and spine arthritis, characteristic deformities of
ruptured wrist extensors, MP severe ulnar deviation, tibio-talar dislocation,
painful effusions of the knee joints, severe morning stiffness, rheumatoid
nodules, and characteristic erosive changes on x-ray, but their rheumatoid
factor is negative, which are you going to believe? That patient has rheumatoid arthritis. The lab tests are not the answer. The same is true for the hypercoagulopathies. When they have it they have it. When a laboratory test is positive, then
your diagnosis and treatment are all the more certain, especially if the
clinical syndromic features were not conclusive by themselves. Knowing the specific faulty chemical can
also help guide therapy depending on which class of chemical is involved
(e.g. prethrombotic gene mutation versus antiphospholipid antibodies). Sometimes the lab confirmation comes not by
way of identifying the culprit, but indirectly by identifying chemical
consequences, such as degradation products of the hyperthrombotic state or
else compensatory changes in other chemicals reflecting up- or
down-regulation in response to that state (e.g. D-dimer might be high, or the
endogenous anticoagulants proteins C, S, and anti-thrombin-3 might be high). What is crucial to appreciate is that hypercoagulable
ulcers are NOT diagnoses of exclusion.
These diagnoses can be made on specific criteria. When lab tests are positive, that always
helps, but history and physical exam are more important than the lab. Remember the essentially pathognomonic
tetrad which is the core of diagnosis for many of these patients: 1
– thrombosis or embolism; 2 – miscarriage; 3
– wound pathergy and ulcers; 4 – auto-immune disorder; (& 5
– personal or family history). It is worthwhile to have standard laboratory panels to order for
suspect situations. These should
include tests for thrombotic species and markers of closely allied or trigger
diseases: factor V Leiden, prothrombin
20210G, antithrombin III, protein C, protein S, APC resistance, fibrinogen,
D-dimer or fibrin degradation products, plasminogen, homocysteine, lupus
anticoagulant, anticardiolipin, cryoglobulins, cryofibrinogen, serum protein
electrophoresis, a screen for connective tissue disorders (ANA and related,
rheumatoid factor). This list is not
exhaustive and is a bit dated. Consult
you own clinical lab for the tests that are available to you. Other useful tests include measures of micro-vascular flow,
including tcpO2, laser doppler, and multispectral surface imaging. Vascular tests of large vessel flow, such
as pvr, ppg, and doppler & duplex are apt to be normal, unless the
patient coincidentally has atherosclerotic arterial disease, or not so
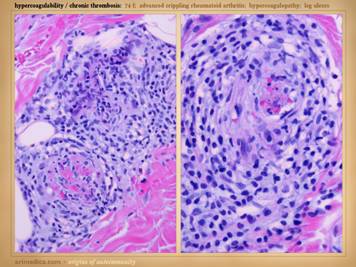
coincidentally has lupus angiopathy of the acral extremities. Histologic exam can be a gold mine of
revelatory changes and positive diagnosis, including findings of: microthrombi and aggregates, minimum acute
inflammation, microvasculopathies, concentric laminations of media due to
repetitive events, vascular fibrosis, vascular stenosis, venous
recanalization, acute neutrophilic vasculitis or peri-vasculitis, and chronic
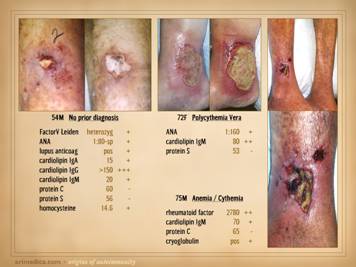
peri-vasculitis with lymphocytes, eosinophils, and plasma cells. Center: chronic thrombosis, vascular occlusion, and
re-organization, in a patient with rheumatoid and proteins C & S
abnormalities (same patient as left-lower on preceding slide). Right: chronic failed wounds and multiple
operations, and persistent skin ulcers following achilles tendon rupture, in
a patient with high fibrinogen, high anticardiolipins, and blind in one eye
due to retinal artery thrombosis. The
ankle is shown left with chronic skin dysplasia and ulceration before
treatment, and right with healed restored skin after diagnosis-specific
treatment, including warfarin anticoagulation. |
|
|
|
|
|
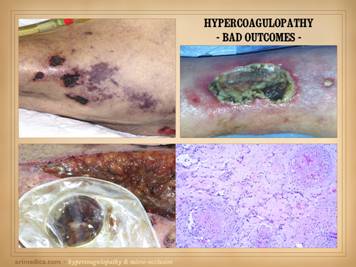
When hypercoagulable patients present with chronic ulcers, some
of their histories can be otherwise quite benign. The wounds and their treatment can be slow,
subtle, persistent, recurring, frustrating, refractory, but a patient’s
general health and well-being are not in any immediate jeopardy – or are
they? Some such patients have
histories of serious prior events, such as blindness due to retinal artery
occlusion, strokes, limb loss from trauma, recurrent pulmonary “emboli”, and
other macro-vascular events. All
hypercoagulable patients have these potential risks. With the appropriate trigger or generalized
inflammatory or hyper-thrombotic state, even the micro-vascular events can
become extensive and life-threatening.
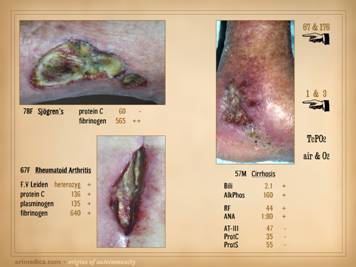
This slide shows three patients who died from these conditions. Left
upper: this patient had heart
surgery, and a week or two after starting warfarin, he developed multiple
non-embolic skin and extremity infarcts.
Peripheral arteries were normal.
Lab studies confirmed low APC resistance and probable factor V abnormality. The events were non-survivable. Lower: this patient had sigmoid resection for a
diverticular colo-vesical fistula.
Bowel necrosis resulted in progressive enterectomy, and with each
procedure, more of the abdominal wall died.
This view shows a necrotic ileostomy and abdominal fascia
infarcts. Lab studies confirmed APC
deficiency. Histology confirmed
diffuse primary micro-thrombosis (i.e. not post-mortem changes, and absence
of significant inflammation pins the thrombosis as the primary event). The events were non-survivable. Right
upper: This patient had refractory
leg ulcers with active progressive infarcts during the period of
observation. Lab evaluation confirmed
primary low proteins C & S. She
died from a stroke shortly after making the diagnosis and planning
treatment. These are non-trivial
diagnoses, and their management must include comprehensive and long term
planning including the role of anti-coagulation. |
|
|
|
|
|
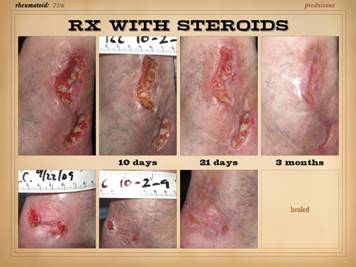
To brighten the mood, let us now look at some of the many
successes that accrue to proper diagnosis and treatment. Always keep in mind the key syndromic
features of hypercoagulability: 1 – thrombosis
or embolism 2– miscarriage 3 – wound
pathergy and ulcers 4 – auto-immune
or connective tissue disorder 5 – personal or
family history Imagine you have seen a patient with a suspicious wound and a
strong history. You try to confirm
your diagnosis with support from the lab.
Next you start the patient on anticoagulants, and then you implement
your plan to close the wound, be it surgery, biologics, wound
pharmaceuticals, or whatever. Problem
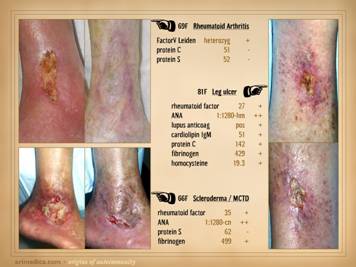
wounds of rapid progression or eons duration now heal. This slide shows three such stories. Left: a 29 year old man with long duration
refractory leg ulcers. History and
profile were suggestive, and the lab confirmed high anticardiolipins – an
antiphospholipid antibody syndrome – and the patient healed just by starting
warfarin. Right upper: a 43 year old
woman, otherwise healthy, but with many years of refractory leg ulcers, and a
history of multiple venous thrombosis and pulmonary embolism or
thrombosis. The lab confirmed low
proteins C&S and low tcpO2’s around the wounds. She healed with warfarin therapy and skin
reconstruction with a regenerative matrix.
She re-ulcerated after she stopped taking warfarin, but then rehealed
after resuming anticoagulation. Right lower: ulceration after skin biopsy in a patient
with cryoglobulins and low protein C (the same patient as “right” on slide
19). She healed with warfarin
anticoagulation and skin restoration with a regenerative matrix. |
|
|
|
|
|
Summary of
hypercoagulopathy The hypercoagulable disorders and ulcers are a major category of
chronic wounds and wound pathology.
They are under appreciated, but overly important. You will not recognize them until you start
to ask the correct questions and incorporate them into your differential
diagnosis. Once a correct diagnosis is
established, then good care and effective outcomes can follow. The treatment of hypercoagulopathic disorders,
wound pathergy, and micro-thrombotic wounds has not been discussed here, nor have
a variety of other issues relevant to the their physiology, pathology, and
clinical management, but much more information on this subject is at the
Arimedica website (arimedica.com). Reviews of
essential subjects - Autoimmunopathy The following slides review immune ulcers and classic immune
disorders. As with the
hypercoagulopathies, this subject is not new, but it is still largely unknown
or under-appreciated by most practitioners.
This section will not be a comprehensive discussion of the subject,
and therapeutics will not be addressed.
The focus will be on issues of anatomical pathology, pathophysiology,
and clinical findings, enough to appreciate sections to follow concerning the
mechanisms of immunopathic ulceration.
To reiterate though, these are a major category of chronic and
pathological CAP wounds, and one of extraordinary importance. As will be developed in subsequent
sections, these are the true diseases of wound healing. |
|
|
|
|
|
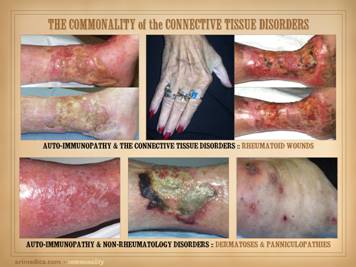
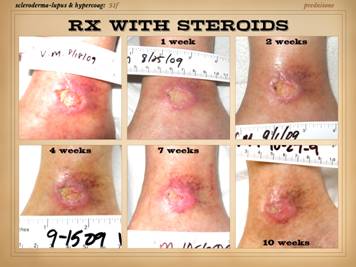
Illustrated on this and the next slide are patients with wounds
and ulcers in association with classic connective tissue and collagen
vascular diseases. All of these
patients were sick to a greater or lesser degree. Some have since lived and done well for
years. Some died after prolonged
chronic disease activity. Some died
acutely from major flare-ups. Some had
a concomitant hypercoagulopathy. None
of them were trivial or easy to manage nor heal. These are bad diseases, hard enough to
manage under any circumstances, but harder yet when complicated by necrosis
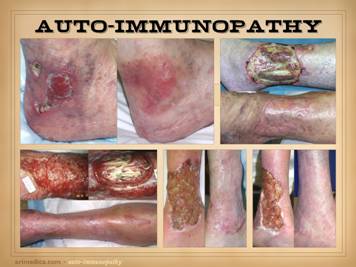
and ulcers. All four of the patients on this slide are shown before and
after treatment of the disease and then skin reconstruction. Left
upper: lupus-rheumatoid-mixed (mctd)
with ulceration due to synovitis and panniculitis. Left
lower: rheumatoid arthritis, with
prototypical rheumatoid ulceration due to synovitis. Right
upper: another prototypical
rheumatoid ulceration due to synovitis.
Right lower: Sjögren’s with chronic panniculitis and leg
ulcers. |
|
|
|
|
|
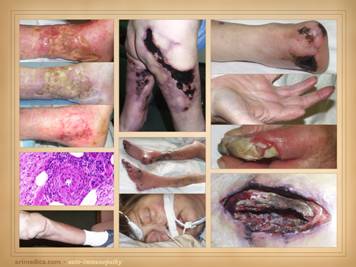
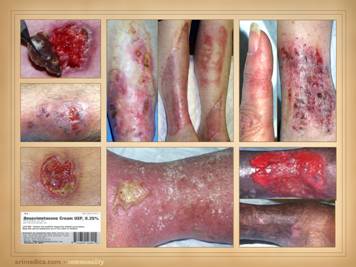
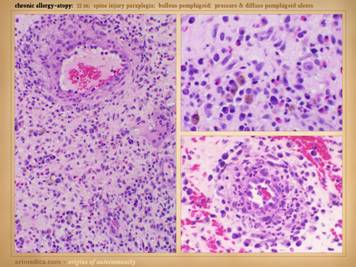
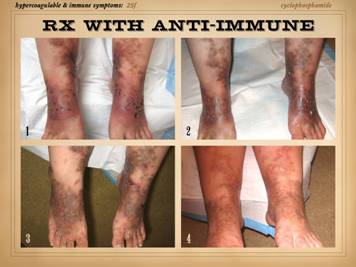
Here are more examples of immunopathic patients and ulcers. Left
upper: Sjögren’s with chronic
panniculitis and leg ulcers. Left middle: (histology only) arteritis with skin ulcers
and necrosis. Left lower: chronic lupus
with multiple wound complications of trauma and surgery. Center
upper: acute lupus with extensive
skin necrosis. Center lower: Behçet’s
with multiple vasculitis, pathergy, thrombosis, and necrosis Right
upper: scleroderma-crest with lupus
angiopathy and multiple skin infarcts and ulcers. Right
lower: rheumatoid arthritis with a
hypercoagulopathy and extensive necrosis following back surgery. |
|
|
|
|
|
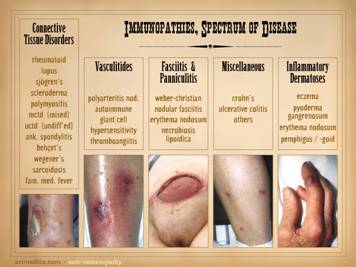
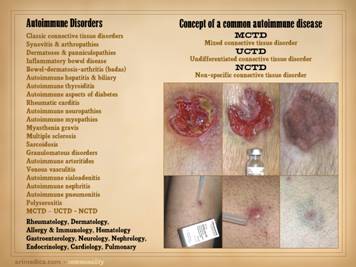
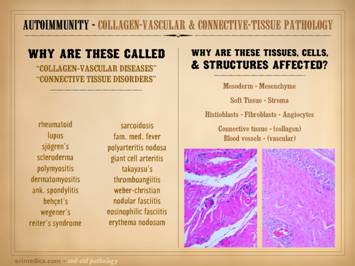
The auto-immunopathies are a spectrum of disease with protean
manifestations. They can affect almost
any chemical, cell, tissue, organ, or system.
Their effects can be parochial and directed against very selective
targets (e.g. Hashimoto’s thyroiditis), or they can be nearly global in
expression (e.g. lupus). There are
various ways to categorize the auto-immune diseases, and this slide gives
several non-exhaustive lists of them: connective tissue disorders, e.g.
rheumatoid, lupus, sjögren’s, scleroderma, polymyositis, ankylosing
spondylitis, behçet’s, wegener’s, sarcoidosis, familial mediterranean
fever; vasculitides, e.g. polyarteritis nodosa, giant cell,
thromboangiitis; panniculitis, e.g. weber-christian, nodular & eosinophilic
fasciitis, erythema nodosum, necrobiosis lipoidica; inflammatory
dermatoses, e.g. eczema, pyoderma gangrenosum, pemphigus, bullous
pemphigoid; miscellaneous, e.g. crohn’s, ulcerative colitis, autoimmune
hepatitis, multiple sclerosis. Once you learn to recognize these diseases and take a thorough
history, you will appreciate that many patients with any of the nominal
primary diagnoses will have a variety of crossover symptoms. Many patients likewise cannot be readily
categorized into any one of the classic named diseases, yet they have strong
features of several of them. In a
certain sense, it is as though auto-immunopathy is a single disease in which,
based on which auto-immunizations and auto-antibodies you get dealt, that
governs the spectrum of signs, symptoms, and complications that you are apt
to have. To account for these
crossover and mix-and-match profiles, patients can be assigned bread basket
diagnoses: mctd (mixed connective
tissue disorder), uctd (undifferentiated connective tissue disorder), nctd
(non-specific connective tissue disorder). How many of these patients and diseases have wound problems? Remember, as a wound practitioner, you will
see patients primarily because of their wounds, and you WILL see all of these
primary diagnoses come through your door.
Conclusions anyone? Images, from left to
right: achilles (Wegener’s granulomatosis); leg (leukocytoclastic arteritis); abdomen (Weber-Christian); leg (Crohn’s); finger (pyoderma gangrenosum). |
|
|
|
|
|
To reiterate, as a wound practitioner, you will see patients
primarily because of their wounds.
When you suspect an immunopathic ulcer or patient, your history and
exam will be directed towards these diseases.
Do this enough times, and it becomes second nature, but until then,
you need a way to think about the multitude and multiplicity of signs,
symptoms, and sequelae that appertain.
Following are a few categorizations.
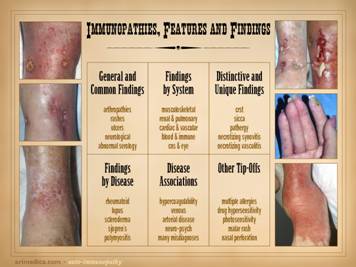
General and common findings:
e.g. malaise, arthralgias and
arthropathies, rashes, sicca, ulcers, neurological, abnormal serologies. Findings
by system: e.g. musculoskeletal,
renal, pulmonary, cardiac & vascular, blood & lymphoreticular, cns
& eye. Distinctive and unique findings: e.g. crst, sicca, pathergy, necrotizing
synovitis, necrotizing vasculitis. Findings by disease: e.g. rheumatoid, lupus, scleroderma,
Sjögren’s, polymyositis. Disease associations: e.g. hypercoagulability, venous, arterial
disease, neuro-psych, many misdiagnoses.
Other tip-offs: common and unusual things, e.g. multiple
allergies, drug hypersensitivity, photosensitivity, malar rash, tendon
rupture, nasal septal perforation. Left upper: rheumatoid arthritis, with acute
panniculitis and multifocal ulceration.
Note the inflammatory-lytic pattern of ulceration, skin dissolution
without infarcted eschar. Left middle: rheumatoid, with multifocal
inflammatory-lytic ulcers. Left lower: lupus or mixed ctd, with atrophie blanche,
dermal scarring from repetitive episodes of connective inflammation. Right
upper: lupus, with suppurative
synovitis. Right middle: scleroderma-crst, with typical features of
fingertip ulcers and necrosis, telangiectasias and sclerodactyly. Right
lower: lupus, with allergic
reaction to common dressing materials. |
|
|
|
|
|
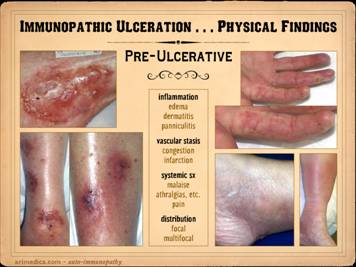
When it comes to examining the skin lesions and wounds due to
these disorders, keep in mind that appearances and features will change as
the injuries and ulcers evolve. First,
you will see a variety of features that occur during the early pre-ulcerative
phases of these lesions, when inflammation and infarction are starting, the
preludes to ulceration. Second, you
will see features characteristic of acute and early ulceration, as the acute
infarctive and inflammatory lesions progress to skin destruction. Third, you will see features of chronic and
late ulceration, as the primary events wind down (or not), and the ulcers
develop gross, histologic, biochemical, and behavioral features of wound
chronicity. This slide shows the
pre-ulcerative changes and features. Inflammatory
signs: Remember, ulceration is
caused by thrombosis-infarction and inflammation-lysis, so what you will see
in advance of actual necrosis and ulceration are the telltale signs of these
states. Because these diseases are
immune and inflammatory in nature, signs of inflammation are usually obvious,
either as dermatitis, panniculitis, cicatritis, inflammatory infiltrates in the
skin, edema, the classic and extended signs of local inflammation, and
systemic inflammatory signs and symptoms.
Vascular stasis signs: Immunopathic inflammation is more apt to
cause primary inflammatory signs, but as will be discussed in detail in subsequent
sections, inflammation triggers thrombosis, the auto-immune disorders
frequently accompany hypercoagulable states, and vessels and vasculitis are
specific targets of auto-immunopathy.
This means that thrombosis, vascular infarction, and stasis are
integral parts of the whole picture.
You are apt to signs of thrombosis and blood stasis, including
congestion and plethora, hemorrhage and skin staining (focal ecchymosis),
cyanotic erythema of these lesions (phlegmasia, as opposed to the scarlet
erythema of inflammation), skin infarcts within these zones, ischemic pain in
the lesions. Systemic signs and symptoms:
These are indicative of the primary disease flaring up, and are due to
a generalized inflammatory state.
These include non-specific general symptoms such as malaise, pain, and
other “flu-like” complaints, along with more focal or tissue specific items
such as arthralgias and stiffness, myalgias, neurolepsy, and the worsening of
other disease- or person-specific symptoms.
Distribution of the
pre-ulcerative lesions and other features that would figure in the assessment
of any dermatosis are also important, such as whether they are single or
multiple, focal or multifocal, blistered-vesicular, macular, papular,
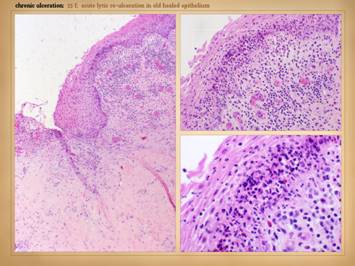
suppurative, eczematous, acneform, desquamative, sclerosing, etc. Left upper: Sweet’s neutrophilic dermatitis with acute
immunopathic neutrophilic abscesses affecting areas of old scar and prior
ulceration. These little abscesses are
the prelude to further focal skin destruction and ulceration. Left
lower: leukocytoclastic vasculitis
(2 different patients) in acute phases of thrombosis, vascular stasis, and
acute inflammation, i.e. the beginnings of infarction and lysis with the risk
of ulceration within days. Right upper: lupus-crst, with sclerodactyly,
telangiectasias, Raynaud’s and angiopathy, prior amputations, contractures,
and eczema. This hand obviously has
high risk based on the primary disease, but the eczema is an acute
inflammatory condition which will trigger the cascade to greater inflammation
and ulceration. Right lower - right: Sjögren’s, with acute panniculitis affecting
the adipose fascias, a common early phase indicator of disease flare up and
potential progression to ulceration. Right lower - left: In this ankle close-up of a similar patient,
note the ring of desquamation, a common indicator of recent acute skin
inflammation, now subsided with treatment, potential ulceration averted. |
|
|
|
|
|
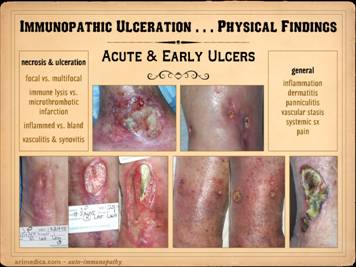
This slide shows the features of acute and early ulcers. Since this is the phase of active
ulceration, you are witnessing the destruction as it happens, as the disease
causes thrombosis-infarction or immunity-inflammation-lysis. You are catching the culprit in the act, so
you are likely to see signs specific to the particular disease, along with
the generic signs of active inflammation and thrombosis, and of active
infarction, tissue lysis, and ulceration. Local findings: These are the features of the necrosis and
active ulceration, in the wounds themselves and in their immediate
surroundings. They may be focal or
multifocal. Multiple or multifocal
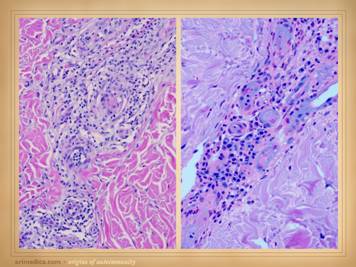
inflammatory-lytic ulceration is usually a dependable sign of an autoimmune origin. There may be inflammatory lysis and
dissolution of tissue versus microthrombotic infarction, which can give
insights as to which diagnosis or mechanism of disease predominates. Whether the periwound is inflamed versus
bland also tends to discriminate thrombo-occlusive lesions from
immune-inflammatory ones. Signs of
vasculitis, synovitis, panniculitis, dermatitis, cicatritis, and even
arthritis and serositis can reveal the autoimmune nature of the problem and
imply which specific disease or syndrome is active. General
and systemic findings: generalized
inflammation, edema, dermatitis, panniculitis, vascular stasis, systemic and
disease-specific symptoms, pain, malaise.
Remember, during these acute phases of ulceration the primary disease
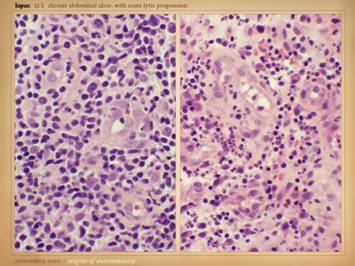
is active, so patients will often have a multitude of symptoms. In the cases shown of early and developing wounds, note the
changes in the ulcers and surrounding tissues. They predominantly show an
inflammatory-lytic pattern of ulceration as opposed to thrombo-infarctive
necrosis. Top left: crst-mctd, dissolution of wound margins,
vascular stasis and cyanosis, acute dermatitis. Top
right: rheumatoid, multifocal
ulceration, periwound inflammation, dissolution of skin without dry eschar,
panniculopathy. Bottom left: rheumatoid,
progressive dissolution of skin and fascias, no eschar, involvement of old
scar, ulceration along tendon sheath and exposure of peroneus tendon. Bottom
center: rheumatoid, multifocal
ulceration, multifocal stasis and cyanosis in advance of infarcts and ulcers,
generalized panniculitis and edema, active necrosis and erosion at skin margins,
no eschar. Bottom right: Sjögren’s,
loss of adipose panniculus, active necrosis and ulceration at margins,
exposure of peroneus muscle (synovitis).
This last example has black necrosis and little inflammation in the
periwound (no erythema nor edema), making this mainly a thrombo-infarctive
pattern of ulceration, implying some type of micro-occlusive pathology. The patient had classic Sjögren’s, but she
also had a very high fibrinogen and low protein C, a good hypercoagulable
explanation for the thrombo-infarctive pattern of the wound. This is the same patient in the same
position (lower right) on the previous slide.
That was her opposite leg, with acute diffuse erythema-nodosum-like
panniculitis, along with generalized signs and symptoms of active
inflammation, immunity, and disease flare up.
As will be shown later, this duality of pathologies – inflammatory and
thrombotic – is common, and many patients will have mixed findings and
features in their wounds, both inflammation-lysis and thrombosis-infarction. |
|
|
|
|
|
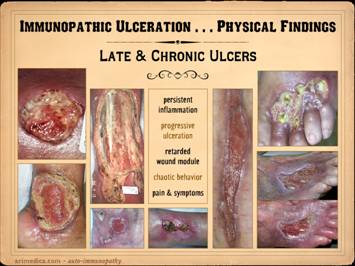
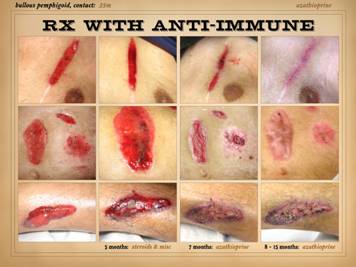
This slide shows the features of chronic and late
ulceration. In chronic ulcers, the
original cause of the disease has subsided, and what is left is just the
anatomical defect. To the extent that
the disease is still active, there may be ongoing slow progression of the
wound or persistence of related findings and general symptoms. To the extent that the disease is affecting
wound repair, or that the ulcer has entered a physiological and pathological
state of chronicity, then healing may be retarded or absent. When looking at the wounds during these
latter chronic phases, what you will mainly see is just a generic chronic
wound. Specific common features to
observe include the following. Persistent inflammation: If primary disease is quiet, then
inflammation may subside, but most such wounds are in a state of chronic
inflammation until treatment brings it under control. Some of it is likely to be a persistent
acute inflammation due to lack of care.
However, acute inflammation is a good stimulus to maintain the
pathological chronic inflammation of the disease, and these wounds rarely
break out of their pathological attractor until both the wound and the
general disease are explicitly treated.
Progressive ulceration: As chronic wounds, most of these ulcers
persist as is, often indefinitely.
However, after prolonged periods of stability they can also get better
or get worse. Many patients describe
prior ulcers which healed spontaneously, typically taking months (and getting
current ulcers healed with treatment is obviously the goal of all of this). To the extent that primary disease or
chronic inflammation in the wound is sustained, there may be slow progressive
necrosis or ulceration. Sudden rapid
progressive ulceration and enlargement is a good indicator of resurgence of
the primary disease. Retarded wound module, mixed wound
module, and chaotic behavior: The
immunopathic disorders have a duality of wound effects. Their afferent effect on the wound is to
make the ulcer. Their efferent effect
is to keep it from healing. There are
very few types of pathology that can arrest the wound module, and active
auto-immunopathy is one of them. For
most patients, wound healing is mixed, both in space over the surface of the
wound, and in time from one observation to another. There may be qualitatively normal
proliferation in some areas. There may
be areas of appropriate suppression of wound healing by acute
inflammation. There can be zones where
the wound module is very weak or inactive due to the primary effects of the
auto-immunopathy, the effects of persistent allied disease states such as
hypercoagulability, and the effects of wound chronicity. There can be intermittent areas of new
ulceration due to persistent chronic disease activity. Even when the wounds look qualitatively normal
at first glance, it is rare that such wounds have quantitatively normal wound
healing kinetics. One of the hallmark
features of wound chronicity is chaotic behavior (as explained in subsequent Parts
1 and 3) in which the wound may wax and wane but never makes any real
progress. Pain & symptoms: There
are only a handful of generic causes of pain (mechanical, neuropathic,
ischemia, cancer, etc.), and inflammation is one of them. Because these ulcers represent an
inflammatory pathology, pain is a common feature. For those who have a concomitant thrombotic
or micro-occlusive disorder, the pains are even worse. Other symptoms of the primary disease, and
secondary symptoms or disabilities related to what area is ulcerated are also
part of the picture of the chronic stages of auto-immune ulceration. In these cases, all wounds are chronic, of long duration, and
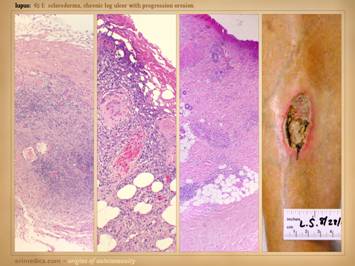
getting some basic topical wound care and treatment for their disease. Left
top: lupus with anticardiolipin
hypercoagulability, zones of granulation tissue, zones without, small active
infarcts at wound edges, persistent inflammation. Left
bottom: rheumatoid, active wound
module of deeper musculoskeletal structures, but no wound healing in subcutaneous
panniculus, active persistent erosions at wound margins, but periwound
inflammation is controlled. Left inner top: rheumatoid with coincidental
atherosclerosis, stable areas mixed with erosive areas, wound healing in
musculoskeletal structures but none in the adipose. Left
inner bottom:
lupus-rheumatoid-mctd, stable wounds, no periwound inflammation,
healing of musculoskeletal structures but not of adipose. Center
lower: polyarteritis, failed
wound module, recurrent acute necrosis.
Right inner: lupus with anticardiolipins, failed
epithelialization, stalled edges, limited contraction, weak
angiogenesis/granulation. Right top: rheumatoid and hypercoagulable, wound
module present at musculoskeletal base but not in subcutaneous fascias, no contraction
nor epithelialization, small surface infarcts even absent periwound
inflammation. Right bottom: rheumatoid,
persistent unchanged wound over a few weeks of observation and care, weak
expression of wound module elements, failed epithelialization and stalled
edges. |
|
|
|
|
|
Whether looking at the prodrome, acute, or chronic phases, many
of the wound features are generic findings of any inflammation, thrombosis,
or ulceration. However, immunopathic
ulcers can have some very distinctive features unlike wounds from other
primary diagnoses. Many of these
reflect specific effects of the given primary disease, such as necrotizing
synovitis from rheumatoid and lupus, and skin sclerosis and calcification
from scleroderma. These features
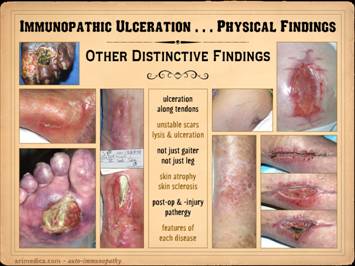
include: ulceration along tendons, due
to synovitis; inflammation, lysis,
ulceration of old scars; ulceration
over small joints, due to synovitis;
inflammation, lysis, necrosis along recent incisions; ulceration of the upper leg outside of the
gaiter area, and ulceration in a variety of other areas; skin atrophy in affected areas, due to
persistent inflammation and proteolysis;
skin sclerosis in affected areas due to scarring after inflammation; vascular changes in skin and
extremity; wound pathergy, necrosis,
and ulceration after injury and surgery;
calcification and ossification in the ulcers or surrounding
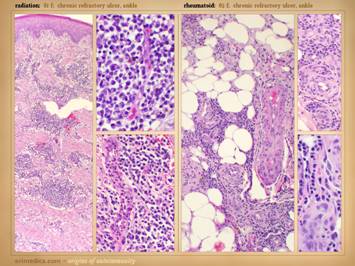
panniculus. Left upper: rheumatoid, ulceration over and into small
joints. Left center: rheumatoid,
ulceration along old scar. Left lower: scleroderma, livedo reticularis, ulceration
along tendons (this ulcer is not under the metatarsal heads). Left
inner: rheumatoid, ulceration
along old scar and tendon. Right inner upper: rheumatoid, ulceration in unusual area
along tendons (thigh, hamstrings). Right inner lower: lupus-mctd, atrophie blanche dermal
scarring. Right upper: rheumatoid,
wound failure of unlikely location (abdomen).
Right lower: rheumatoid and hypercoagulable, wound
failure of unlikely location (forearm), ulceration along muscle and tendon,
necrosis around staples. |
|
|
|
|
|
In examining and diagnosing immunopathic ulcers, or indeed
ulcers of any cause, it is crucial to observe what is not there. Consider for instance the significance of
pulses as an indicator of macro-arterial disease. Absence of pulses implies arterial
insufficiency. An associated wound
could be due to arterial disease, or else an ulcer of unrelated cause (e.g.
rheumatoid or trauma) might heal slowly, and further evaluation is
needed. However, if good pulses are
present, then macro-arterial disease is ruled out. If a suspect rheumatoid or lupus wound has
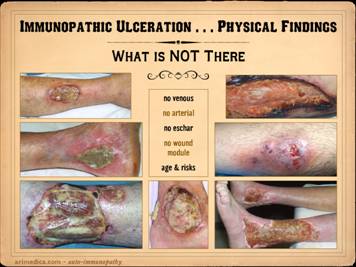
good pulses, then arterial disease is NOT there. What else is not there in immunopathic
ulceration? No arterial: no signs of
vascular disease, no claudication, no change in pulses or pressures or
dopplers. No venous: no signs of
venous disease, no dermato-liposclerosis, no hemosiderin pigment changes, no
venous varicosities nor reflux, no chronic edema nor phlegmasia. No
eschar: immunopathic ulceration is
more apt to be inflammatory-lytic in nature, not thrombo-infarctive, so dry
eschar is not apt to appear. No wound module: as will be explained in later slides, the
immunopathic disorders are the diseases of wound healing, and therefore the
proliferative wound module which does the healing is apt to be flawed or even
absent. Seeing wounds in which
underlying anatomical structures remain pristine visible for months, devoid
of angiogenesis and fibroplasia, is not an everyday occurrence, but nor is it
rare, usually occurring in severe metabolic wrecks or with the auto-immune
disorders. Age & risks: various
ages and diseases are apt to cause certain types of wounds. For example, greater age has greater
implications for arterial disease, and diabetes and neuropathy cause very
characteristic syndromic wounds such as malperforans ulcers. It is important to observe that a patient
or wound does not have those pathognomonic features of other disorders, nor
that there is a discrepancy between actual findings and demographic
expectations, all of which tend to rule out competing diagnoses. There are of course, in examining individual patients and
wounds, many exceptions to these generalities. The net of all observations is what is most
important, not just any single parameter.
In these cases, observe what is absent: Left
upper: rheumatoid, no edema, no
pigment changes, no liposclerosis, no chronic dermatitis. Left
middle: rheumatoid, no edema, no
pigment changes, no liposclerosis, no chronic dermatitis. Left
lower: rheumatoid, no edema, no
pigment changes, no liposclerosis, no chronic dermatitis, no subcutaneous
fascias (exposed muscle and tendon indicative of tenosynovitis). Center:
rheumatoid, no edema, no pigment
changes, no liposclerosis, no chronic dermatitis, no subcutaneous fascias
(exposed ligaments and tendons consistent with synovitis). Right
upper: severe acute lupus, no
peri-wound inflammation, no edema, no wound module (i.e. no healing). Right
middle: ulcerative colitis and
pyoderma, no generalized panniculitis or dermatitis, no involvement near
ankle, no pigment changes. Right lower: Sjögren’s, no pigment changes, no
generalized dermatitis, no generalized liposclerosis, no signs of arterial
disease, no venous varicosities. |
|
|
|
|
|
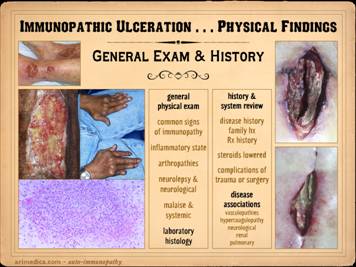
In examining and diagnosing any ulcer or patient, it is crucial
to assess the entire person, their history, review of systems, and general
exam. Oftentimes the diagnosis will
flow directly from the history or spectrum of symptoms. It is important for wound practitioners to
become versed in the signs and symptoms of all of the relevant primary
diseases that underlie chronic and pathological ulcers, be it arterial ,
diabetes, hematological disorders, rheumatological diseases, and everything
else. The more you ask about these
relevant histories, the more you run an inventory of signs and symptoms, then
the more automatic it becomes. Patient intake must include history and examination for all
facets of these diseases, but the following are key components or interesting
issues that will be frequent findings in a wound practice and are often key
features that establish the diagnosis on first encounter. History
& system review: disease
history, family history, treatment history, complications of trauma or
surgery. General physical exam:
common signs of immunopathy, signs of a general inflammatory state,
arthropathies, sicca, neurolepsy & other neurological changes, rashes,
malaise & systemic symptoms. Disease associations: vasculopathies, hypercoagulopathy and
hematological, neurological, renal, pulmonary, miscarriage, venous disease,
dermatoses, inflammatory bowel disease and other organ-specific
auto-immunopathies. Laboratory: serologies, hypercoagulation studies,
vascular assessments, wound histology. New patients with these diseases often have some extremely
common or else distinctive profiles on initial wound exams. The following few examples immediately give
away the auto-immunopathic cause of the ulcer, whether or not they had a
prior diagnosis. These profiles are
important to recognize and hard to overlook, unless the history is sloppy,
inexpert, or just ignored. Notice that
these profiles are based on general history and exam alone, not on anything
about the ulcers themselves. [1] – One very common profile is the
patient with rheumatoid or whatever who has had a recent adjustment in his
drugs. This is often someone doing
extremely well, enough that his chronic daily prednisone dose was lowered,
typically from 10 mg to 7 mg, or from 7 mg to 5 mg. Arthralgias, stiffness, and malaise flare
up, with leg panniculitis or synovitis causing skin ulceration. [2]
– Another common profile is the patient who comes with a typical leg
ulcer. His hands have advanced
rheumatoid degeneration. He has
complaints of symmetrical polyarthralgias and morning stiffness. Are you smarter than a fifth grader? How could anyone have missed this? However, the patient has been explicitly
told that he does not have rheumatoid, and he has been denied treatment
because his rheumatoid factor and other serologies were negative. [3]
– Another profile, not so common but of immediate importance, is the patient
who comes in for a leg ulcer or whatever wound. Examination is hampered by a state of
neurolepsy, being apathetic, disoriented or disengaged, psychomotor retarded,
and just not “being there” or “out of it”.
The patient might also have some history of “seizures” or “MS” or
other central neuropathy refractory to treatment. He is plethoric, has malar rashes, and
signs of arthritis, synovitis, panniculitis, or sicca. This patient has lupus (or Sjögren’s or
rheumatoid or Behçet’s or mixed-ctd or whatever), and he needs steroids and
other treatment right now. It is most important to realize that the wound practitioner is
frequently going to be the first to make the diagnosis of the underlying
problem, or to correct a misdiagnosis that the patient has been given. If a rheumatoid patient goes to an
orthopedic surgeon’s office for an arthritis problem, odds are the patient
already has an established diagnosis, and if not, the orthopedist is likely
to recognize it. If a patient with
lupus nephritis is referred to a nephrologist for renal failure, odds are the
diagnosis is already known, or else the nephrologist will make it. For wounds, patients are referred to
somebody because there is a hole in the skin that is freaking somebody
out. Patients rarely come with any
insight as to the diagnosis. Even if
they have well established diagnoses of rheumatoid, polymyositis, or
whatever, their other doctors have rarely drawn the connection to the
wound. Sadly, when these patients show
up to many self-designated “wound specialists” who really are not, the ulcers
and the overt history of rheumatoid are never connected. However, there are also many patients with
wounds due to autoimmune and connective tissue disorders where the primary
diagnosis has never been made.
Sometimes the patients have subtle signs and symptoms of the disease. Sometimes they have gotten so used to
chronic symptoms of malaise and arthralgias that they hardly recognize that
they are systemically ill. Sometimes
new patients are seriously sick with underlying disease out of control. The patients may know that they feel lousy,
but they often have no other clue that there is a systemic problem, even in
the face of serious symptoms. To get
the wounds better, the disease has to be diagnosed and treated, and that
means you! Left upper: lupus-mctd, symptoms of neurolepsy,
arthralgias, stiffness, sicca, malaise.
Left lower: rheumatoid, typical hand changes, typical
histologic changes of chronic vasculitis.
Right upper & lower: two patients with rheumatoid and
hypercoagulable states, both with necrosis of the wound following back
surgery, demonstrating the types of serious trauma and surgery complications
that can happen with these diseases. |
|
|
|
|
|
Finally, the intake history for people with immunopathic wounds
must include a treatment history. Even
if the patient is not understanding of their own history and disease, their
medication list may betray that they are already being treated for connective
tissue disorders. Treatment history
may reveal that disease flared up and ulcers appeared when certain therapies
were started, stopped, or adjusted.
Failed and favorable responses to prior therapies are valuable in
confirming diagnosis and planning further treatment. The responses to treatment may be
considered as follows. Correct responses: steroids, anti-immune, or anti-inflammatory
drugs were given to treat disease; the
patient or wound had a positive response to such drugs. No
responses: failed or ineffective
therapies for the wound or disease;
multiple failed therapies with different agents or at different
times; wound surgery failures such as
skin grafts which did not take. Adverse responses: disease or wound flare-ups due to
treatment, such as lowering steroid doses;
wound pathergy and wound complications of surgery; atopic dermatitis or atopic vasculitis or
other allergic responses to treatments (common in many auto-immune patients
who have multiple drug allergies). Contrary responses: inflammation, wound infarction, and
progressive ulceration from treatments meant to improve the wound. Contrary wound responses can occur with
cytokines (e.g. pdgf, anti-tnf-α), living cell
therapies (engineered living skin equivalents), and immune competent
chemicals (e.g. monoclonal antibodies). Left upper: rheumatoid, 52 failed skin grafts (yes, he
counted them). Left lower: lupus-mctd,
resurgent ulceration after pdgf therapy.
Left inner: rheumatoid, healing induced after systemic
steroids and anti-rheumatoid drugs (plus typical topical wound care). Right
upper: rheumatoid and
hypercoagulable, inflammation arrested by steroids and warfarin, but wound
healing not induced. Right lower: Crohn’s disease of skin, existing ulcers
healed after intralesional steroids, and new lesions prevented from
ulcerating by prompt steroid injection. |
|
|
|
|
|
Summary of
autoimmunopathy and the connective tissue disorders The “rheumatoid” and immunopathic ulcers, due to the classic
connective tissue or rheumatological diseases, are a major category of chronic
wounds and wound pathology, under appreciated, but overly important. They are common, generally easy to
recognize and diagnose, but only if you are aware of them and conduct the
proper patient interview. The wound
practitioner will often be the first one to make the diagnosis of a systemic
connective tissue disorder. Successful
treatment of the ulcers is contingent on proper diagnosis and treatment of
the primary disease. |
|
|
|
|
|
Auto-immunopathy and related wounds come in a variety of
flavors, and practitioners from different specialties will have a different
taste of the problem. Rheumatologists
will see the auto-immunopathies from the point of view of the collagen-vascular
and connective tissue disorders. This
is a diverse point of view, but there are other auto-immune diseases that may
not be so likely to show up in a rheumatologist’s office, such as
inflammatory bowel disease, auto-immune thyroiditis, pemphigus, and multiple
sclerosis. There are generalities and
commonalities of these diverse conditions, and they all make problem wounds,
and they all show up in a wound practice.
We will start by looking at the immune mediated dermatoses and
panniculopathies, a large category of illness most likely to be seen by a
dermatologist . . . and in a wound practice.
Dermatoses are common and are the face value stuff of
dermatology and dermatology textbooks.
Many skin problems will show up in a wound practice, and having some
familiarity with dermatology is important.
There are a few generic categories of dermatological disease that you
will see commonly, such as the eczemas, pemphigoid, atopic, and other
inflammatory dermatoses. Panniculitis and the panniculopathies are crucial to understand.
The “panniculus” refers to the subcutaneous adipose fascias. There are two subcutaneous layers, Camper’s
and Scarpa’s fascias, plus areas or layers of areolar adipose scattered
throughout the body. These fascias are
just another connective tissue, a generic stroma of fibroblasts and
angiocytes playing host to adipocytes. The adipose panniculus is the prime target of a lot of
autoimmune attack. Many of the leg
ulcers you see start as inflammation in the panniculus, and ulceration occurs
as the overlying skin dies due to thrombosis of its supply vessels
underneath, or due to lysis from contiguous inflammation. While auto-immunopathy can result in
primary dermatitis, primary synovitis, primary fasciitis and ligamentitis,
and just about primary anything else, it is important to realize the crucial
role that the adipose fascias have in being the prime target and wellspring
of many of the skin ulcers that occur with immune and inflammatory diseases. As for so many of the dermatoses and related conditions, there
is a gargantuan nomenclature of the inflammatory dermatoses and likewise for
the panniculopathies. If you read a
dermatology textbook on the subject, the list of names and diagnoses will take
many columns. However, it is easy to
see that many are duplicates, or “blind-men-and-the-elephant” differing
perspectives on the same thing, or descriptive names based on physical
features rather than pathology (cf. “atrophie blanche” for dermal scarring),
or old names from bygone eras when the relevant physiology and pathology were
not understood. Some of the more
common or relevant disorders will be listed on slide 30. Regardless of all of the descriptive names
and legacy nomenclature, we are talking about a central pathology in which
immune, allergic, and inflammatory events are turned on against the host,
specifically the skin and the adipose and areolar fascias. These disorders also have a duality of
adverse effects, first causing damage and then impairing the ability of the
body to repair that damage. Top left: long standing severe rheumatoid with
chronic leg ulcers. Many rheumatoid
ulcers occur over tendon sheaths, a manifestation of primary synovitis, but a
more diffuse primary panniculitis with secondary dermatitis and ulceration is
also common. (See also the photos
slide 20 left upper, and slide 22 bottom center.) Top
right: a similar patient with long
standing rheumatoid disease with primary panniculitis and diffuse leg
ulceration. Although the patient had
all of the symptoms and criteria for the diagnosis of rheumatoid, her
arthritis, arthralgias, and joint damage were relatively mild. However, the panniculopathy was severe,
illustrating how the “skin” and wound manifestations of the disease are
sometimes the most rampant manifestation of disease, sometimes even the only
active aspect of disease. Bottom left: simple postural stasis leading to secondary
panniculitis and an eczematous dermatitis,
Bottom center: pyoderma gangrenosum in a patient with
subtle lupus-like symptoms. Bottom right: necrotizing panniculitis of the thighs in a
patient with undifferentiated or mixed connective tissue disease. The subject of postural stasis needs some comment, since this is
one of the most common and commonly misunderstood and misdiagnosed entities
that a wound practice has to deal with.
This occurs due to the accumulation of dependent edema. It is common in people who sit and do
nothing all day with their legs down (typically obese sedentary older
people), in people with heart or lung disease who must sit up at all times
and sleep that way, and in the dependent part of the obese overhanging
panniculus of abdomen and thigh. Edema
and fluid stasis leads to leukocyte stasis which then triggers acute
inflammation. Panniculitis is the
primary event, with secondary eczema of the skin. These people simply need good compression
or other edema control, very short term anti-inflammatory therapy, and
non-specific wound and skin care. This
care is extraordinarily simple in principle, and 100% effective, but can be
exasperatingly hard to implement or maintain in many of these people. These are the patients who are often
mislabeled as “cellulitis” (an essentially meaningless term to begin with)
and treated with antibiotics and other irrelevant things while never getting
any of the correct care, turning a simple benign easy-to-fix problem into
chaos and complications. |
|
|
|
|
|
Here are examples of inflammatory dermatoses and
panniculopathies causing skin ulcers.
Expect to see any and all of this in a wound practice. Left upper: pyoderma gangrenosum in an otherwise
healthy young woman (primary inflammation at dermal-hypodermal
boundary). Left middle: pyoderma
gangrenosum in a patient with ulcerative colitis (primary inflammation at
dermal-hypodermal boundary). Left lower: Crohn’s disease, primary lesion in the skin
(primary inflammation at dermal-hypodermal boundary). Center
upper: necrobiosis lipoidica in an
otherwise healthy woman (primary inflammation in the subcutaneous
panniculus). Center lower: postural
stasis (primary inflammation in the panniculus with secondary
dermatitis). Right upper: simple eczema
and atopic dermatitis (primary dermatitis).
Right lower: bullous pemphigoid or eosinophilic
dermatitis (primary dermatitis). |
|
|
|
|
|
Dermatology is a vast subject, and only a small part of it
involves wound related issues.
Likewise, wounds are a vast subject, and not all wounds involve the
skin, and only a portion are due to primary dermatoses and
panniculopathies. Yet dermatology and
wound practice have a very important area of intersection. These are the ulcerative dermatoses and
panniculopathies, and they are almost exclusively of
immune-allergic-inflammatory origin. There are many ways to mix, match, and rearrange them in a table
of nomenclatures, A few major
categorizations are given below. What
is common to all is that they represent a state of auto-sensitization or
auto-immunization. They affect all
ages. They have a spectrum of extent
and severity. They may or may not be
associated with some other major syndromic disorder (e.g. lupus or
inflammatory bowel disease). They are
all responsive to anti-inflammatory, anti-allergic, or anti-immune therapies,
and their steroid responsiveness is the cornerstone of acute and chronic
treatment. This list is far from complete – it is just a sampling of common
diagnoses that will be seen regularly in a wound practice, all within the
realm of the ulcerative and inflammatory dermatoses and panniculopathies. Dermatoses: eczema, pyoderma, pemphigus, pemphigoid,
Sweet’s (neutrophilic dermatosis). Panniculopathies: Weber-Christian (and other lobular
panniculopathies), erythema nodosum (and other septal panniculopathies),
lipomembraneous panniculitis, necrobiosis lipoidica, nodular fasciitis,
eosinophilic fasciitis. Collagen-vascular and connective tissue
disorders: lupus-rheumatoid-Sjögren’s,
poly-dermatomyositis, scleroderma-crst, Behçet’s, inflammatory bowel
disease. Vasculitis: leukocytoclastic,
polyarteritis nodosa, venous vasculitis.
Miscellaneous: uncategorized, drug eruptions, contact
eruptions, intertriginous dermatitis. |
|
|
|
|
|
The rheumatologists see one aspect of autoimmune disorders. Dermatologists see another. In fact, doctors from almost any specialty
are going to see their own facet of autoimmune and inflammatory
diseases. The following is a list of
confirmed or putative auto-immune disorders.
If you were a primary physician making referrals, they would go to
many different specialties, e.g. rheumatology, dermatology, allergy &
immunology, hematology, gastroenterology, neurology, nephrology,
endocrinology, cardiology, pulmonary.
As a wound practitioner, you are in the one specialty that will see
them all. Here is a sampling of these disorders, the list constrained by
what would fit on the slide, in no particular order: classic connective tissue disorders,
synovitis & arthropathies, dermatoses & panniculopathies, inflammatory bowel disease, bowel associated dermatosis-arthritis
syndrome (badas), autoimmune hepatitis & cholangitis, autoimmune
thyroiditis, autoimmune aspects of diabetes, rheumatic carditis and rheumatic
fever, autoimmune neuropathies, autoimmune myopathies, myasthenia gravis,
multiple sclerosis, sarcoidosis, granulomatous disorders, autoimmune
arteritides, venous vasculitis, autoimmune sialoadenitis, autoimmune
nephritis, polyserositis, mixed connective tissue disorders (mctd-nctd-uctd). Keep in mind that many of these patients and disorders will have
a mix-and-match set of signs and symptoms, and these crossover profiles have
necessitated the use of generic terms like mixed and undifferentiated
connective tissue disorder. The more
thorough you are in taking a history and inventory of symptoms, the more you
will find. In a sense, it is as though
auto-immunopathy is just as single generic disease, presenting different
profiles, symptoms, and sequelae based on which specific antibodies appear
and which specific cells or tissues get targeted. |
|
|
|
|
|
To summarize these two short reviews, remember the
following. The hypercoagulable
disorders and ulcers are a major category of chronic wounds and wound
pathology, under appreciated, but overly important. The “rheumatoid” & immunopathic
diseases and ulcers are also a major category of chronic wounds and wound
pathology, likewise under appreciated but overly important. The autoimmune disorders are a broad class of disease that will
be seen, in one form or another, by physicians from almost all
specialties. They are manifest in a
variety of common and distinctive syndromic patterns, and they are thus
classified by an accepted nosological nomenclature (e.g. major names like
lupus or rheumatoid or eczema).
However, these nomenclatures are artifice and an alias for the real
pathologies that underlie a state of auto-sensitization or
auto-immunization. They all tend to
have a variety of crossover features or symptoms, and in a sense they are
conceptually all a single disease, all a form of MCTD - UCTD. All carry some risk of wound problems, and
all such diagnoses will be seen in a busy wound practice. |
|
|
|
|
|
The connections
between autoimmunopathy and connective tissue disorders The upcoming sections will make the connections between
autoimmunopathy, connective tissue disorders, and wound healing. When we think about the “connective tissue
diseases”, it is all about the autoimmune disorders that are typically within
the purview of the specialty of rheumatology (along with the many autoimmune
disorders seen by other specialists). Why
are these diseases called “collagen-vascular” and “connective tissue”
disorders? Why are they related or due
to autoimmune states? Why are they not
related to some other general class of pathology? Why are there no common diseases of the fascias,
connective tissues, and general stroma related to metabolic alterations or
genetic deficiencies? To explain the
autoimmune origins of the connective tissue disorders, we will look first at the
effects of autoimmunity directed against the stromal connective tissues, i.e.
the tissue pathologies that result from autoimmune sensitization against “collagen”
and “vascular”. After that, we will look
at why autoimmunopathy arises and is directed against the connective tissues. |
|
|
|
|
|
Why are these called “collagen-vascular diseases” and
“connective tissue disorders”? First,
let us clarify some basic terminology about the soft and connective
tissues. The connective tissues are the
general stroma or support structure of the body. They are all basically a structural matrix
of protein fibers. The predominant chemical
is collagen, and this matrix is the basic fabric of all connective
tissues. The matrix has to be made by
some sort of cell, and that is the fibroblast. However, nothing lives without substrate
supply, and this depends on a vascular distribution system – blood vessels –
created by angiocytes. These are the
two constituent cells of the general connective stroma of the body –
fibroblasts and angiocytes – period.
These structures can play host to other cells, such as adipocytes, but
the fibrous stroma of the body depends on just fibroblasts and
angiocytes. The term “histioblast”
will also be used here to denote tissue forming progenitor cells that spawn the
fibroblasts. Recall that all of these
cells are part of the mesenchyme, the tissues derived from the embryonic
mesoderm. Illustrated are two
prototypical examples of basic stromal or connective tissue. On the left
is scar from a healthy trauma wound in early phases of maturation. On the right
is normal muscular fascia. These
tissues appear slightly different because, depending on the specific tissues
and circumstances, the collagen architecture varies in expected and
predictable ways. However, they appear
more similar than different, and that is because they are formed of just two
structures, blood vessels in collagen matrix, which are made by just two
cells, angiocytes and fibroblasts. |
|
|
|
|
|
The stromal tissues, engineered by the mesenchymal cells of the
mesoderm, predominantly histioblasts-fibroblasts and angiocytes, are composed
of collagen and connective proteins, traversed by blood vessels and vascular
structures. Diseases which affect the
mesenchymal stroma therefore explicitly affect these cells and
structures. It is predominantly the
autoimmune disorders which do this.
Why are these cells and structures the target of
auto-immunopathy? To begin the answer,
this slide shows what that targeted pathology looks like. Bottom
left: acute leukocytoclastic
vasculitis. This is arteritis in its
acute phases, with intense neutrophil infiltration with necrosis and myxoid
degeneration of the vessel wall.
Vessels are clearly an explicit target of this event. Bottom
right: rheumatoid synovitis,
likewise with neutrophilic acute inflammation and myxoid changes in a tissue
that is nothing but loose fibrous stroma with fibroblasts. Top
left: chronic vasculitis or
peri-arteritis (from a patient with long-standing ulcers and infarcts due to
a combined coagulopathic and auto-immune disorder mainly consistent with
polyarteritis nodosa). Neutrophils
have disappeared, and instead, chronic inflammation has ensued, consisting of
lymphocytes, plasma cells, and eosinophils.
Note how the pathology is confined to the vascular locus, without
inflammation in the surrounding connective matrix. Note also the chronic thrombosis in the
damaged vessels. In anticipation of
explanations soon to come, ask yourself this crucial question: what came first, the thrombosis or the
inflammation? |
|
|
|
|
|
The consequence of auto-immune attack is to kill cells and
trigger inflammation. Inflammation is
a destructive process by intent, so when triggered, there is progressive lysis
or damage of the affected tissues.
Inflammation also begets wound repair, so fibrosis and related
consequences of the wound module also appear, i.e. scar. The net effect is acute and chronic damage and
scarring of mesenchymal and stromal tissues, including their derivative
structures such as the fascias, vessels, and musculoskeletal structures. This slide shows those anatomical and
clinical effects that result form auto-immunopathy and inflammation of the
collagen-vascular connective tissue stroma.
Top left: fibrosis develops in blood vessels after
vasculitis, leading to stenosis and obstruction; these angiogram shows
paradigm “lupus angiopathy” which occurs most commonly in lupus and
scleroderma. Bottom left: a
cross-section view of a dermal artery from a scleroderma patient showing the
mural fibrosis and stenosis; this patient’s skin was also highly fibrotic
from repetitive inflammation and scarring of the dermis, confirming both
“collagen” and “vascular” targets of his disease. Bottom
middle: prototypical late stage
rheumatoid hands; these deformities are the biomechanical consequences of
destruction of tendons and joint ligaments, destroyed by chronic active
inflammation triggered by autoimmunity targeted against synovium. Top
middle: earlier stage rheumatoid
synovitis during a flare up; the leg has become ulcerated because of global anti-connective
tissue effects affecting more than just synovium. Top
right: atrophie blanche, a classic
morphological feature of skin in some of these disorders, representing areas
of residual normal skin interspersed with dermal scarring due to repetitive
dermatitis-fibrositis. Bottom-right: “string-of-beads” ulceration characteristic
of autoimmune synovitis; in the hand, rheumatoid synovitis is apt to cause
tendon rupture, whereas in the lower extremity, it is apt to lyse skin; the
upper photo is a lupus patient during acute phases of synovial suppuration
and skin ulceration; the lower photo is a rheumatoid with chronic ulceration
after the synovitis flared and has now subsided. |
|
|
|
|
|
The autoimmune disorders can affect nearly any cell or tissue in
the body – e.g. immune thyroiditis, hepato-biliary disease, inflammatory
bowel disease, hidradenitis, etc. When
it is the mesodermal-mesenchymal connective and vascular stroma of the body that
is the target of auto-immunopathy, then the tissues affected are those of the
fascias and musculoskeletal system, as seen on the last slide. Depending on which antibodies or cells and
tissues are predominantly targeted, the patient may have a syndromic set of
signs and symptoms that fit into standard nosological categories. Thus, synovitis dominant disease is likely
to fit diagnostic criteria for rheumatoid arthritis. Dominant involvement of the adipose fascias
is apt to be recognized as Weber-Christian, erythema nodosum, and related
panniculopathies. Muscle involvement
may prompt a diagnosis of polymyositis.
Serositis dominant disease might be called lupus or
Weber-Christian. Complications of
fresh wounds and old scars are apt to fit with lupus. Other distinctive events, such as uveitis,
spondylitis, secretory adenitis, urethritis, cerebritis, central vasculitis,
mediastinitis, etc. will all betray certain syndromic diseases and
classifications, such as Reiter’s, ankylosing spondylitis, Sjögren’s,
Behçet’s, Wegener’s, Takayasu’s, etc.
Many patients of course will have mix-and-match findings necessitating
the use of “mixed”, “undifferentiated”, and “non-specific” to describe the
connective tissue disorder. Regardless
of what the primary diagnosis, predominant target, or syndromic signs are,
there is one thing that is common to all of these diseases: because stromal connective and vascular
tissues are involved or targeted, therefore skin ulcers, wound pathergy, and
similar soft tissue events are common. |
|
|
|
|
|
We have just looked at the tissue pathology and clinical effects
that result from the autoimmune collagen-vascular & connective-tissue
disorders. We can now answer why these
are called “collagen-vascular diseases” and “connective tissue disorders”. They are so called because the immune
events and their active agents (antibodies, lymphocytes, etc.) target the
mesenchymal cells (fibroblasts & angiocytes) which constitute the stroma
(connective matrix and blood vessels) of all tissues. The gross anatomical pathology and the
clinical sequelae of these diseases are due to (1) active inflammation
damaging connective and other tissues, such as acute synovitis and
panniculitis, (2) anatomical changes resulting from that destruction, such as
tendon and ligament rupture, joint deformity, and skin ulcers, and (3) the
effects of scar, such as vascular stenosis and stiff joints. The photo is from a chronic non-healing leg ulcer in a
patient with active rheumatoid. This
is a view just below the surface (which is just out of view at the top),
showing the topmost plasma protein stratum and the subjacent angio-attraction
layer. On the left half is an
area of plasma proteins with neutrophils and angioid cells – a normal mix of
normal wound “fauna”. Vascular
organization and density is far too low for a normal wound, aminoglycans are
probably hypodense, and there is edema, all of which make this an abnormal
pathological wound, typical of many chronic immunopathic wounds. The cells are not organizing properly, but
the cell mix itself and their density within the plasma are largely
correct. On the right half is a
vascular locus, a cluster of organizing new vessels which is intrinsically
correct, as would be expected in the angio-attraction and angio-organization
layers. However, the architecture of
the vascular locus is masked and disorganized by a dense infiltrate of
inflammatory cells confined to the locus.
This infiltrate contains some neutrophils, but it is predominantly
plasma cells (antibody producing B-lymphocytes). Why would these immune mediator cells be
attracted to the new vessel?
Understanding why gets to the heart of why collagen-vascular
auto-sensitization and the connective tissue diseases occur. |
|
|
|
|
|
So far, you have been asked to accept on faith and common basic
knowledge that auto-immunity is the cause of the connective tissue
disorders. We have just illustrated
the anatomical pathologies that result from the auto-immune state and thus
why they are called collagen-vascular diseases. The next big question – a two-parter –
is (1) what are the origins of
autoimmunity in the first place, and (2) why does autoimmunity target these
tissues? The origins of rheumatoid, lupus, and auto-immunopathy have had
various theories and debates.
Infections, repetitive trauma, and other inflammatory states have all
been implicated. None of these is entirely
correct, but what they all share is a state of chronic inflammation. What you will see here is how a chronic
inflammatory state of any primary cause can lead to auto-immunization. Start by considering two other auto-immunopathies
where the origins of the auto-immunity is understood. (1) Rheumatic fever and rheumatic heart
disease occur because strep throat, scarlet fever, or other streptococcal
disease induces immunity to streptococcal antigens. Key to this is the streptococcal PARF
antigen (“peptide associated with rheumatic fever”), which binds to human
collagen. When immune cells become
sensitized to the bacterial antigen, they also develop immunity to the
conjoined collagen, and now you have an auto-immune connective tissue
disorder. (2) Spina bifida and
myelomeningocele have a high incidence of allergy to latex. Now how odd is that? A meningocele exposes neuroectodermal
tissues to the general circulation and mesenchyme, where inflammatory cells
can meet them. The CNS is rich in a
variety of phospholipids, so some type of low level sensitization to these
chemicals occurs. Latex, raw rubber,
is a micellular suspension of isoprene monomer globules suspended in
phospholipid membranes – phospholipids, the same stuff as in the CNS. Exposure to latex probably acts as a
secondary sensitizer, a booster shot if you will for auto-immunity which is
already present to some degree. Latex
then further acts as a trigger for acute responses. The response is more allergic than immune,
but it is nonetheless an example of sensitization developing to
auto-antigens. In the rheumatic fever
case, auto-immunization occurs because human collagen is in the way, at the
wrong place at the wrong time, caught up in the melee as immunity develops to
an exogenous immunogen. In the spina
bifida case, auto-immunization occurs because non-mesenchymal antigens
normally hidden from the immune system become exposed. How does this relate to the common
connective tissue disorders and the problems that frequent a wound practice? Consider now another disease, hidradenitis suppurativa. This is a type of acne that affects
apocrine glands. Everybody gets overly
focused on the suppurative abscesses in the obstructed glands, but the real
problem that obstructs the glands is a lymphocyte and antibody mediated
autoimmunity directed against the glands.
How does this develop? The
glands are epithelial structures, filled with degenerated cellular
debris. These materials normally are
on their side of their basement membrane, NEVER exposed to the mesenchyme
underneath. When the glands inflame
and rupture, epithelial endocellular debris and sebaceous chemicals become
exposed to and get intermingled with the mesenchyme. This means that macrophages and lymphocytes
can now “see” them, allowing immune sensitization to occur. Similar events presumably explain the
uveitis that occurs in the eye (as with Behçet’s and Reiter’s), and perhaps
as well antigens in the secretory lacrimal and salivary glands (Sjögren’s),
and so on. So, how is it that the body
develops immunity to these chemicals?
Is it that they are “visible” normally non-immunogenic chemicals that
caught up in the fracas of some acute inflammatory event, as occurs with
streptococcus-induced rheumatic fever?
Or is that they were sequestered chemicals, never meant to meet a
lymphocyte in real life, but barriers fell as occurs with spina bifida and
latex cross-over allergy? For the
diseases that are apt to show up in a wound practice, we can find strong
compelling evidence of both mechanisms. Throughout this discussion, we keep talking about auto-immunity
and antibodies. Antibodies to
what? The easiest way to appreciate
this is to look at what antibodies we can measure readily. The clinical lab has various tools to
assess auto-immunity, mainly antibody assays.
Anti-nuclear antibodies are the most prevalent, and they serve as a
basic screen. If positive, further
testing can reveal others. The
table on the right shows some tests and panels available from a large
commercial lab. Listed on the left
are a greater variety of auto-antibody assays that can easily be
ordered: actin (smooth muscle), antinuclear antibodies, cardiolipin, centromere,
chromatin, cyclic citrullinated peptide (CCP), dsDNA, endomysial, histone,
interleukin-2 receptor, Jo-1(histidine-tRNA ligase), liver kidney microsome
(LKM-1), mitochondrial, neutrophil cytoplasmic (ANCA), perinuclear ANCA,
ribosomal P, ribonucleo-protein RNP, scleroderma (Scl-70), sjögren’s SS-A Ro,
sjögren’s SS-B La, Sm, Sm-RNP, tissue transglutaminase (ATA).
Do you see a pattern? Look closely. These assays are for auto-antibodies
against nuclear chromatin and DNA, centromeres, endoplasmic reticulum and
ribosomes, mitochondria, selective enzymes, etc. The commonality is that these auto-antibodies
are all directed against intra-cellular structures, some intra-cytoplasmic,
and some intra-nuclear – aka “anti-nuclear antibodies”. |
|
|
|
|
|
This slide will illustrate some of the conditions and mechanisms
whereby nuclear and cytoplasmic auto-immunization occurs. Left
lower: a normal specimen from a
healthy active open wound. The
angiocytes of the blood vessels are all hyperplastic and partly disaggregated,
a normal effect of angiogenic cytokines in the proliferating wound, i.e.
normal and healthy. There is stasis
and migration of polymorphonuclear leukocytes – completely normal behavior
after any injury or thrombosis – completely proper reactive inflammation. In addition to well-formed poly’s, you will
also see dark spots that look like the size and shape of individual lobes of
a neutrophil’s nucleus – because they are.
This is “nuclear debris”, aka “poly dust”. As neutrophils conclude their business and
do the apoptosis thing, nuclear remnants and other cellular debris are left,
waiting to be cleared by macrophages or other degradative cleanup
mechanisms. The more intense the
inflammation, the more concentrated the loose nuclear or cytoplasmic material
becomes. In specimens with intense
necrosis and suppuration, there is always a generalized purple basophilia
reflecting the high load of loose endocellular material. Simply put, normal inflammation results in
the release of endo-cellular materials into the interstitial medium. Right
upper: a specimen from an unhealthy
wound, a patient with polyarteritis nodosa (the photo shows new vessels and
other elements of the reactive wound, not the primary pathology with acute
suppurative inflammation around old vessels).
There is vascular disorganization with fibrin exudates, heavily
infiltrated with poly’s, and a substantial amount of poly dust. The vessels are the target of acute
inflammation, but unlike a short term inflammatory state after trauma, this
inflammation is repetitive and sustained.
Compared to healthier wounds, the load of free cellular and nuclear
debris is quantitatively increased and also more sustained in time. This increases the exposure of autologous
materials that are normally sequestered inside cells and hidden from the view
of antigen processing mechanisms. This
abnormally heightened exposure increases the chances of antigenic
recognition. Right lower: a specimen
from a long standing pressure ulcer with areas of new necrosis. This type of wound is presumably
non-pathological, just a matter of pressure and trauma, but nonetheless
chronic. The vessels and stroma are as
expected in this upper (plasma-inflammatory) stratum of the wound, and there
are poly’s and poly dust as you would expect.
Large monocyte-macrophages are also present, likewise as
expected. However, there is also
immunogenic inflammation – eosinophils and plasma cells – not very dense in
this particular view, but there nonetheless.
Why would the body be acting as though it is immunized and making
immunoglobulins against itself in this chronic seemingly benign wound? You can start to see how auto-immunization
occurs. All of that nuclear debris is
freely exposed in the tissue. What
should be protected antigens sequestered inside cells are being exposed where
immune processing cells (macrophages and lymphocytes) can pick them up. The more the material, the longer it is
exposed, the greater the chance of recognition and sensitization. And not only are the sequestered antigens
at risk (like happens with spina bifida and latex), but even chemicals
normally exposed in the area risk being picked up and carried along on the
immunization ride (like for rheumatic fever).
And what normal chemicals or structures risk being carried along? Whatever happens to be there. Those associated with the stromal matrix or
its resident cells and structures – fibroblasts, angiocytes, vascular
structures, and connective structures. |
|
|
|
|
|
We have just looked at how intracellular debris becomes exposed
during conditions of necrosis and inflammation. The more sustained the injury and
inflammation, then the more prolonged the exposure of potential antigens, and
the more likely that immune recognition and auto-sensitization might take
place. On this slide, we will review
the mechanisms of immune recognition and sensitization, events contingent on
the presentation of potential antigens
to lymphocytes. As we have already reviewed, two types
of potential auto-antigens can occur. (1) Exposure of occult and sequestered antigens,
in which materials that are normally “invisible” to the mesenchyme and
interstitium become exposed and accessible to immune processing cells
(macrophages and lymphocytes). These
include: the endocellular debris of
acute necrosis and inflammation, leading to intra-cytoplasmic antibodies; endonuclear debris of the same origin,
leading to anti-nuclear antibodies; cell
and tissue specific debris, such as with hidradenitis; and antigen cross-reactivities such as
occurs with latex and spina bifida. (2) Opsonization, haptenization,
inverse-haptenization of open antigens, in which small or normally
non-antigenic chemicals combine with other chemicals or structures to create
larger immunogenic intermediaries that induce sensitization to the primary
chemical. This is the mechanism of auto-immunization
with parf-induced post-streptococcal rheumatic carditis and rheumatic
fever. It might also be the mechanism
of cross-over sensitization in the uveitis of Reiter’s and similar syndromes. In the wound, the abnormal bindings to
“exposed chemicals” that could lead to sensitization presumably occur against
small degradation fragments of connective matrix that has been chewed up in
the course of injury and inflammation. Left upper: an image of a severe lumbar gibbus in a
patient with spina bifida and latex allergy.
It is a good illustration of how the privileged domain of the CNS is
violated. The cord is interrupted, and
neural elements intermix with mesenchyme, making unique CNS chemicals such as
the phospholipids “visible” to the macrophages and lymphocytes that govern
immunization. The potential for
auto-immunization gets worse in principle if there is also a focus of injury
and inflammation which recruits and concentrates chronic and inflammatory immune
cells, such as a pressure ulcer over the gibbus, as this patient had. Left lower: a patient with hidradenitis
suppurative. Chronic inter-gluteal
disease is shown, representing 30 years of sustained inflammation. This is another example of sequestered
antigens (in this case epithelial and sebaceous) being exposed to the
mesenchyme, where auto-immunization can occur. Not only was the disease responsive to
anti-inflammatory and anti-immune drugs, but as discussed further on slide 53,
this patient also had significant rheumatoid symptoms which cleared when the
disease was excised and then healed (middle and lower images). It should be clear by now that various
primary injuries or pathologies can expose sequestered antigens or else
create novel antigens, thereby leading to immune recognition and
auto-sensitization. How does this
recognition and sensitization occur?
The process starts with the raw materials, the generic debris of
neutrophils and acute inflammation, and the site-specific debris local cells
and matrix. These materials must now
be processed and packaged for presentation to the immune system, the
lymphocytes. Who is the intermediary
that does the processing and presentation?
Macrophages. Remember, the
afferent role of the macrophage in the wound is as a phagocytic cell that has
the task of clearing debris.
(Macrophages laden with debris are also called histiocytes.) As debris is digested, detoxified, or
whatever is happening in the bowels of the macrophage, certain chemicals will
be packaged for export to lymphocytes.
The more sustained the injury or acute inflammation, the more likely
that endocellular and other autogenous debris will eventually be flagged as
“foreign” and presented to a lymphocyte for immunization. Once auto-immunization does occur, the
involved area will develop a cohort of “chronic inflammation”, infiltration
with immune and allergic cells – lymphocytes, plasma cells, and eosinophils. Right top: a chronic abdominal wound form a patient
who is other wise healthy and free of any clinical auto-immune syndromes or
symptoms. In the inflammatory and
upper aminoglycan layer of the wound, large mononuclear cells and neutrophils
share the stage. The large cell on the
left is more of a native pre-transformation monocyte, whereas the cell to its
right is a post-transformational macrophage.
Inside that macrophage is a nuclear lobe from a neutrophil. This is a good illustration of how
inflammatory debris is processed, and the means by which auto-immunization
against nuclear antigens and the formation of anti-nuclear antibodies can
occur as the acute inflammatory state is sustained. Right middle: a chronic abdominal wound from a young
patient with a diffuse chronic bullous pemphigoid and extensive multi-focal
ulceration. The lesions all have a
complex mix of acute (generic reactive) and chronic (immune)
inflammation. Within one small field
we can see acute inflammation (poly’s and nuclear debris) and chronic
inflammation (lymphocytes, plasma cells, an eosinophil). There are also macrophages, and possibly
mesenchymal repair cells (the two smaller spindle cells are most likely
migratory angiocytes). The large
central cell, stippled by intracytoplasmic debris, sits next to all of this,
closely hugging a plasma cell. While
the plasma cell is a committed antibody producer, not involved in new
sensitizations, it can be seen here how debris-processing macrophages will
easily get a chance to exchange something with lymphoid cells. Right lower: another view from the same patient, in
which cells are denser, with the same admixture of multiple populations. Debris laden histiocytes (the brown color
is presumably hemosiderin from the processing of erythrocytes) are nestled
against lymphocytes and plasma cells.
It is the “perfect storm” of events for auto-immunization. Sustained acute inflammation creates greater
production, density, timewise exposure, and histiocytic processing of occult
intra-cellular antigens. A greater
admixture of “chronic inflammatory” immune cells attests to auto-immunization
having already occurred, but also increases the likelihood of uncommitted
lymphocytes being presented with new auto-antigens. Being a wound, the “locals” are angiocytes
and stromal cells and their products (angiocytes, fibroblasts, vessels,
matrix), and their exposure in this process increases the likelihood that
their own debris will become part of the immune exposure and auto-sensitization,
leading to stromal auto-immunization. |
|
|
|
|
|
The origins of
autoimmunopathy and connective tissue disorders via other primary diseases We have just looked at the autoimmune nature of the connective
tissue disorders. We looked at why
they are called “collagen-vascular diseases” and “connective-tissue
disorders”, because of the effects of autoimmunity directed against the
stromal connective tissues. We then
looked at why endocellular antibodies and autoimmunopathy arise, and how they
can become directed against the connective tissues. So far the focus has been on auto-immunity
and collagen-vascular disease per se, although the last two slides gave some
hint of how all of this might relate to chronic wounds. Our next step, before squarely relating
this to wounds, is to look at how auto-immunopathy and the connective tissue
disorders arise in the first place from some other primary underlying
disease. The concept here is that some
other primary disease creates the conditions of sustained injury, necrosis,
and inflammation which lead to the recognition of sequestered and
endocellular antigens, thereby leading to auto-immunization and the resulting
connective tissue disorders. |
|
|
|
|
|
This slide starts a series that demonstrate a fascinating and
crucial connection in this story – the relationship of hypercoagulable
disorders to auto-immune disorders.
Recall in our basic review of hypercoagulable states and ulcers that
there is a recognizable, nearly pathognomonic tetrad / pentad that nails the
diagnosis of a hypercoagulable disorder:
(1) thrombotic or embolic event, (2) miscarriage, (3) wound pathergy event,
and (4) a connective tissue disorder, with (5) either a personal or family
history. Why the connection between
these two major disease categories?
Left: a woman with scleroderma-crest.
History and wound behavior suggested a hypercoagulable state,
confirmed by laboratory, and she ultimately died from pulmonary embolism or
thrombosis. Note the significant
multifactorial markers of both autoimmunopathy and hypercoagulability. Right: a man with clinically active lupus
with multiple wounds and wound complications due to minor trauma and surgery. Wound specimens confirmed thrombi, the lab
confirmed antiphospholipid antibodies, and he healed promptly with
warfarin. Why did both patients have
unequivocal evidence of both disorders? |
|
|
|
|
|
You might think that the two patients on the preceding slide
just had an unhappy coincidence of dual diseases. You might, until you see this and the next
few slides. On this slide are three
patients presenting with obvious pathological ulcers, including active
necrosis, inflammation, and progressive ulceration. One of them had no prior documented
history, and the other two had confirmed histories of polycythemia or other
blood cell disorders. Laboratory
evaluation using customary panels for autoimmune and hypercoagulable
disorders showed evidence of both in all three of these patients. None of them had a clinical history of
autoimmunopathy or connective tissue disease, but on symptom inventory and
review of systems they had a variety of typical complaints such as arthralgias
or sicca syndrome. The profile of the
54 year old man is especially noteworthy.
In addition to autoimmune markers, he has a dual type of coagulopathy:
factor V Leiden indicates an intrinsic pre-thrombotic hypercoagulopathy, and
he also has a strong elevation of immune procoagulants (antiphospholipid
antibodies), both lupus anticoagulant and anticardiolipin. You cannot cheat on a gene test - factor V
Leiden is an inborn error, a built in “pre-thrombotic” or intrinsic hypercoagulable
disorder. So isn’t one coagulation
defect enough for one person? Well,
not for him, but why – why would he then also get auto-immune procoagulant
antibodies? And why would markers of
collagen-vascular auto-immunity accompany this? No, it’s not coincidence. |
|
|
|
|
|
More of the same, three more examples of mixed laboratory
findings in patients with clinical profiles that indicate dual disease, both
coagulopathic and immunopathic. On the
left, 2 patients with a priori established clinical diagnoses of a connective
tissue disorder (top = rheumatoid, bottom = scleroderma-mctd). In both, the behaviors of the ulcer implied
a coagulopathic origin as well, and this was confirmed in the lab. Both healed with a customary program of
anticoagulation and skin reconstruction with a regenerative matrix. The 69 year old rheumatoid patient had
overt crippling clinical disease.
However, similar to the case on the preceding slide, she also had
factor V Leiden, a built in genetic “root” of the system around which
everything else must be explained. On
the right is a prototypical acute pathological wound in a patient with no
antecedent clinical disease or diagnosis.
The features are more thrombo-infarctive than inflammatory-lytic, but
there are elements of both, both grossly and by laboratory assay. |
|
|
|
|
|
And another one . . .
These three patients had clinical histories of active immunopathy plus
acute ulcers behaving more thrombo-infarctive rather than
inflammatory-lytic. The duality of the
problem was confirmed on laboratory profiles.
Note that the rheumatoid patient has markers that cannot be mistaken –
clinical and pathological rheumatoid for which she just had her back operated
on, and genetically inborn factor V Leiden.
(Yet another such case. Notice
that plasminogen, a natural thrombolysin, and protein C, a natural
anticoagulant, are both elevated, a typical reflex up-regulation of these
compounds in response to a continuing state of thrombosis.) I could keep showing you more of the same
thing, lots more . . . Are you
convinced now that these are not coincidences? In our clinic, we have standardized sets of laboratory blood
tests that we order in working up these ulcers and diseases, a “rheumatoid
panel” and a ‘”coag panel”. For a long
time, we have been drawing both panels on our primary coagulopathic patients,
so the “data” and experience are more thorough for those patients. Only recently have we started drawing
“hypercoag” panels on our primary “rheumatoid” patients, and as we do more,
the real values for this set should become clearer. There are also plenty of patients who come
with ulcer profiles and clinical histories that make both sets of disease
clear and obvious from the outset, and it is not surprising that their
laboratory workups have mixed markers.
Other patients have ulcers and profiles that are vague or confusing,
suggestive but not overtly diagnostic of either category. The very reason we started drawing both
sets of laboratory panels in the first place is that so many of these
patients have wounds with both features, thrombo-infarctive and
inflammatory-lytic, and who also, upon thorough history and exam, have a
suggestion of both sets of disease. I have yet to do a formal retrospective (nor prospective) data
analysis of our experience with these patients and profiles. However, having paid attention to these
issues for the past 14 years, and having observed them in hundreds of
patients, I have a general sense about the incidence of these
correlations. Remember, all of these
patients come to our practice because of the wounds, not because of a
characteristic rheumatological or hematological complaint. (1)
For patients presenting with a primary hypercoagulopathy, i.e. where
the wounds and history and laboratory profile are strongly “hematological”,
more thrombo-infarctive in nature, then the incidence of laboratory
cross-over with positive auto-immune markers seems to be well over 50%,
perhaps as high as 85%. (2) For
patients whose clinical profiles are strongly “rheumatological”, with overt
history or symptoms, and with inflammatory-lytic ulcers, the presence of
hypercoagulable markers seems to be in the range of about 25% to 50%,
depending on the primary rheumatological diagnosis. |
|
|
|
|
|
Why? Why is there such a
strong association of autoimmunopathy with the hypercoagulable states? Why do so many of these patients have
markers of two major categories of disease?
The clinical and laboratory findings may be occult or overt, subtle or
dramatic, but they are there when you look for them. The explanation is not so mysterious, and
it begins with the basic mutual interaction of thrombosis and
inflammation. To begin, let us observe
histologically the wound and inflammatory anatomy of patients with this
duality of hypercoagulability and auto-immunity. Here are two slides from two patients with hypercoagulable
disorders. The specimens came from
wound biopsies or debridements, meaning that there was already established
ulceration. What do you see? Both show a vascular locus, a zone of
vessels and angioid tissue within a dermal or connective matrix. Within the vascular locus, vessels have
chronic thrombi and reorganization.
The specimen on the left shows migratory spindle shaped
disorganization of the angiocytes consistent with a state of chronic
inflammation and wound healing. Both
specimens have diffuse infiltration of the vascular locus with chronic
inflammatory cells. Recall that
whereas acute inflammation is neutrophilic, chronic inflammation shows
predominantly lymphocytes, plasma cells, and eosinophils. The left specimen is infiltrated with
almost pure lymphocytes. The right
specimen is infiltrated with mostly all plasma cells and eosinophils. There are few neutrophils, and the
surrounding collagen matrix shows normal fibroblasts without inflammatory
changes. Why are these vessels in a
state of chronic immunogenic inflammation? The short answer is that the hypercoagulopathy is chronically
making microthrombi. These can cause
repetitive micro-infarcts and inflammation.
With persistent necrosis and inflammation, there is an increasing load
of cellular debris and increasing activities to clean it up. Lymphoid cells are not part of normal acute
wound healing, but with chronicity they begin to appear. All of this increases the possibility (and
eventually the likelihood) that macrophages will have a chance to present
autogenous materials to lymphocytes which in turn will get confused into
thinking that these materials require an immune response. Normal healthy incidental wound healing is not subject to these
dynamics nor to auto-immunization. The
key to the onset of this state of chronicity is some sort sustained injury
that perpetuates inflammation and forces the admixture of cells that normally
should not “see” each other.
Hypercoagulability is one cause of this, but there are others, as
discussed below. First though, let us
take a look at the dynamics of wounds and tissues under conditions of
hypercoagulability or other sustained primary injury. |
|
|
|
|
|
Let us reiterate the message of the last slide. In certain patients with refractory wounds,
why do we see vessels in a state of chronic immunogenic inflammation? Why do we see these patients have clinical
and laboratory profiles indicative of both hypercoagulability and
auto-immunopathy? It happens because
hypercoagulopathy chronically makes microthrombi; these cause repetitive micro-infarcts and
inflammation; persistent necrosis and
inflammation create a sustained load of cellular debris and macrophage
cleanup activities; this raises the
chances that lymphocytes (not a normal part of acute inflammation and wound
healing) will become sensitized to normally sequestered endocellular
antigens; once sensitized,
auto-immunity can develop to the vessels and other stromal elements which
were caught in the middle of these repetitive events. The stromal elements go from being passive
victims of repeated incidental injury-infarct-inflammation to being active
targets of sustained injury-infarct-inflammation. Furthermore, the induced auto-immunity and
chronic inflammation leads to locally thrombogenic conditions, triggering new
thrombosis in the same area in the hypercoagulable patient, thereby
perpetuating the entire process. Normal patients with normal wounds do not develop stromal
auto-immunization. The difference
between the normal state and the chronic and pathological wound is that
something, such as a hypercoagulable state, is sustaining the injury,
thrombosis, necrosis, and acute inflammation that is required to perpetuate
these events. We are all used to
thinking about inflammation as either acute (neutrophilic) versus chronic
(lympho-plasmacytic), but now it is time to understand a third state: sustained
acute inflammation. This is the
bridge between incidental injury with one-shot responses (acute inflammation)
versus self-perpetuating auto-pathological states with auto-immunization
(chronic inflammation). To understand the significance of sustained acute inflammation,
both as its own pathophysiological entity, as well as its effects on impaired
wound healing, we will begin by looking at the fundamental relationships and
mutual interactions of thrombosis and inflammation. Recall the quintessential roles and
functions of inflammation and thrombosis.
How is an injury recognized?
How is it cleaned up? How is
the repair process started? There are
several pathways of injury recognition, and one of them is platelet
activation and thrombosis. Once these
events occur, they then initiate inflammation, so the body can handle the
defenses, do damage control, and then clean up. Thus, thrombosis triggers
inflammation. However, inflammation
also creates a milieu that promotes thrombosis via prothrombotic chemicals,
leukocyte and platelet trapping, morphological and chemical changes in
vessels, changes in blood viscosity, etc.
Thus, inflammation triggers thrombosis. They trigger each other. This complex non-linear system is
self-amplifying. In the case of a
one-shot incidental injury, such as trauma, this thrombosis-inflammation
coupling ensures a swift ramp up of defensive changes, but then the process
subsides and settles, paving the way for repair. However, when there is some sort of
repetitive or sustained injury, then new thrombosis and inflammation keep
getting triggered, keeping the process alive.
For the technically savvy, it is very much analogous to an oscillatory
system in mechanics or electricity. A
one-shot trigger in a spring or vibrator circuit may oscillate briefly as it
falls back to zero, but if you keep kicking in a bit of energy at the
resonant frequency, enough to replenish internal energy losses, then you can
sustain the oscillation and make cool things work, like a radio or a
clock. Trauma induced
thrombosis-inflammation is a one-shot.
Thrombosis-inflammation triggered by chronic sustained thrombotic or
immune disorders and activities keeps the system running. When acute inflammation is reactive and one
shot limited, it is proper and works for the defense of the host. When inflammation becomes sustained,
self-perpetuating, then chronic, that is improper and works to the detriment
of the host. What triggers abnormal or sustained thrombosis and
inflammation? Active auto-immunopathies
do this. Primary or intrinsic
hypercoagulopathies also do this. So
too angiopathies and panniculopathies, i.e. disorders of the stromal
structures which thereby also trigger thrombosis and inflammation. These elements have a variety of complex
interactions, but at their core are the mutual interdependence of thrombosis
and inflammation. If there is some
factor which can sustain or re-trigger either of these, then the
thrombosis-inflammation cycle can become sustained. And what is caught in the middle? The health or not of the host tissue – infarction
and lysis, necrosis and ulceration. If
the thrombotic events predominate, then thrombo-infarctive necrosis is more
apt to be seen. If the inflammatory
events predominate, then lysis and ulceration are more apt to be seen. It should be no surprise though that many
wounds and patients will have features of both events, seen grossly,
histologically, and in the laboratory. While thrombosis and inflammation have a mutual interdependence,
they do not automatically trigger and re-trigger each other and thereby enter
a self-sustaining loop by default.
This is the difference between acute inflammation and sustained acute
inflammation. Look again at the
logarithmic timeline of injury response events. An acute self-limited injury occurs. It triggers thrombosis. Thrombosis dynamics are a one-shot, a swift
ramp-up then a decay, subsiding because no further trigger nor provocation
sustains it. Thrombosis serves as an
integrator that at some threshold or level then triggers acute
inflammation. By the time inflammation
is strongly on the rise, inflammation is waning. The two events barely have any timewise
overlap. Inflammation is itself a
one-shot, which in turn stimulates the wound module, with these two events
likewise having little overlap. Normal
inflammation is thrombogenic in principle, but absent other associated
perturbations or amplifications, it is a relatively weak thrombogen. Thus, inflammation, which feeds forward
into wound module, does not ordinarily feed backward to re-trigger or
perpetuate thrombosis. However, when
some other sustained injury, primary thrombosis, or primary inflammation is
present due to unrelated, antecedent, coincidental primary or independent
pathology or trauma, then re-triggering and sustentation can start. Once the thrombosis-inflammation mutual
loop starts to revolve, then there will be abnormal feedbacks into the
injury, thrombosis, and inflammation cusps of the overall timeline. As discussed in Part 1, slide 32, chaotic
dynamics begin, and the wound cannot converge (and may even diverge, getting
worse due to thrombo-infarction or inflammation-lysis). A primary hypercoagulable disorder is one of those a priori
underlying conditions that can create the sustentation that perpetuates the
thrombosis-inflammation link and drives a wound into chaos. Normal one-shot inflammation may only be
weakly thrombogenic, but in a patient where the thrombosis trigger is overly
sensitive, the threshold for clotting abnormally low, then “weakly
thrombogenic” may be enough. Of
course, new thrombosis, and also the associated necrosis, will then
(strongly) trigger new inflammation. It
should be clear by now that simple acute (one-shot) inflammation and
sustained acute inflammation are dynamically very different states or
attractors. Sustained acute
inflammation is what happens when there is repetitive or persistent action of
any of the normal triggers of acute inflammation (trauma, allergy, immunity,
infection, thrombosis). Under the
right circumstances, the mutual dependence of thrombosis, inflammation, and
related elements can turn “repetitive action” into auto-sustentation. How it is that sustained acute inflammation then leads to the third
state, auto-immunization and chronic inflammation, will be explained on the
next several slides. What is key to
understand is that wound or stromal auto-immunity and chronic inflammation do
not occur after simple one-shot injury-inflammation-healing wound
dynamics. They occur only after a
period of sustained acute inflammation, a necessity for
auto-sensitization. The following are
the characteristics of these three inflammatory domains. Acute
inflammation: one-shot
dynamics; there is minimum necrosis
and minimum exposure of sequestered antigen;
there is minimum, hardly any macrophage-lymphocyte admixture. Sustained
acute inflammation: dynamics are
of repetitive, persistent, unresolved primary injury, or else some other
disorder or condition that can trigger
injury-thrombosis-necrosis-inflammation events; sustained events increase the load of
cellular and nuclear debris; as events
drag on, there are increased amounts and times of exposure of debris and cell
elements; increased time and
increasing levels of lymphokines raise the risk of lymphocyte appearance and
admixture; the whole milieu now increases
the chances of macrophage-lymphocyte admixture and interaction with
presentation of local auto-antigens that have appeared due to the sustained
thrombosis, necrosis, and lysis. Chronic inflammation: a new population of cells enters the mix, lympho-plasmacytic
infiltrates, meaning that auto-sensitization against stromal elements has
occurred; this is a state of intrinsic
pathology that perpetuates injury and altered dynamics, and it can no longer
be averted just by breaking the cycle of thrombosis-inflammation. |
|
|
|
|
|
This photo was shown on slide 45. It is from a 75 year old woman with a
hypercoagulable state. It shows
chronic thrombi and reorganization within a vascular locus which is also
densely infiltrated with lymphocytes.
This reflects more than just local stromal auto-immunization – this
patient also has chronic active overt crippling rheumatoid arthritis. We have now looked at many similar cases
where patients with problem wounds had clinical and laboratory features of
concurrent hypercoagulability and auto-immunity. On slide 34 we asked what came first,
thrombosis or inflammation? We are now
ready to fully answer that question, but first a final clarification, about
the meaning of chronic inflammation. “Chronic inflammation” has two connotations. (1) There is a vernacular everyday use of
the phrase which implies that inflammation has simply been there a long
time. The implication is that this is
generic or non-specific acute neutrophilic inflammation, and that it has
become sustained or persistent. This
loose and inexact concept simply assumes that inflammation is inflammation,
and that when it is there too long, it is ipso facto chronic. (2) Chronic inflammation has a stricter
pathological connotation of inflammation with lymphocytes, plasma cells, and
eosinophils. These are latter
populations of cells which are not present in acute inflammation. However, by using the term “chronic
inflammation”, as opposed to “immune” or “lympho-plasmacytic inflammation”,
people tend to get the idea that “chronic inflammation” is chronic simply
because it has been there a long time, that lymphocytes and plasma cells must
be just the relief team for the long suffering and overworked neutrophil
team. This is not true. These two classes of cells share a
commonality that they are both involved in host defense, but they otherwise
have distinct functions, and lympho-plasmacytes have no role in acute injury
and wound healing. This is why we need the concept of “sustained acute
inflammation”, to describe conditions when neutrophilic acute inflammation
has become repetitive, persistent, enduring, and perpetuated, as opposed to
the short-term one-shot dynamics of simple acute inflammation. Chronic inflammation is an entirely
different third state which occurs when injury and acute inflammation have
been present a long time (“chronic” in the vernacular sense, but “sustained”
in our nomenclature) leading to infiltration with immune mediator lymphoid
cells (“chronic inflammation” in the jargon of pathology). So, which came first, thrombosis or inflammation, the
hypercoagulable state or the autoimmune disorder? The answer is that the hypercoagulability
and thrombosis came first. Lupus,
rheumatoid arthritis, Sjögren’s, and related auto-immune connective tissue
disorders are the consequence of factor V Leiden, prothrombin 20210G, and
similar primary hypercoagulable states.
As described below, the hypercoagulable disorders are not the exclusive
cause of these connective tissue disorders, but for patients with a primary
hypercoagulopathy, the relationship between the hyperthrombotic and
auto-immune states is direct and causal.
The pathway to auto-immunization depends on the sequence of (1)
injury, thrombosis and acute inflammation being perpetuated by some other
abnormal factor, leading to (2) sustained acute inflammation, which creates
conditions which permit auto-sensitization against stromal elements, which in
turn leads to (3) chronic lymphoid inflammation which can perpetuate the
wound even after the injury-thrombosis-inflammation loops of acute disease
have been arrested, and which furthermore can cause systemic anti-stromal
pathologies, i.e. the connective tissue disorders. On the preceding slide, we focused on how some type of
pathological state can lead from acute inflammation to sustained acute
inflammation. Here we will now focus
on how that state of sustained acute inflammation leads to auto-sensitization
and chronic lympho-plasmacytic auto-immune inflammation directed against
native cells and tissues. Autoimmunity
occurs when the immune system “sees” sequestered or novel auto-antigens that
it should never have seen. The
potential auto-antigens are either endo-cellular and nuclear debris and other
occult & sequestered antigens that are exposed by inflammation (including
cell lysis, apoptosis, phagocytosis, etc), or else exposed matrix chemicals
modified and opsonized or inverse-haptenized by inflammatory chemistry. This potentially antigenic debris does not
cause any effect in normal one-shot inflammation, because antigenic load is
small, and the admixture of mononuclear and other acute inflammatory cells
with immunogenic lymphoid cells is limited by the times and spatial distributions
of events in a normal healthy wound.
However, with sustained acute inflammation, there is a persistent
increase in cell kill and debris, antigenic load, and admixture of different
cell populations that are normally separated.
Specifically, macrophages are processing the debris. Increasing loads of debris means increasing
chances of processing a specific chemical, and then presenting it to an ever
increasing admixture of lymphocytes.
Macrophage-lymphocyte interactions are limited in one-shot injury and
acute inflammation, but sustained acute inflammation increases admixture, in
both time and space within the wound, and the risk of chance sensitization
increases. Auto-immunity occurs when
that type of antigen processing and presentation “scores”, allowing a
lymphocyte to develop sensitivity to that antigen. The key to wound auto-immunization, i.e. sensitization to
normally safe or sequestered auto-antigens, is the sustained acute inflammation.
This is the state that leads to the spatial and timewise “stirring” of
the wound, causing admixture of cells and events that are normally separated
during normal healthy one-shot responses in a normal incidental wound. This process can occur with any condition
which can re-trigger, perpetuate, or chaoticize the wound by provoking or
repeating out-of-sequence or out-of-turn injury-thrombosis-inflammation. Once the wound or patient is auto-immunized,
protean clinical sequelae can ensue, anything from persistent local wound
pathology and impaired healing to grand systemic morbidity for which lupus or
mctd (mixed cvd-ctd) is the index disease. Hypercoagulopathies are one of the obvious, dominant, and easy
to identify causes of this auto-immunization, leading to both wound problems
and systemic connective tissue disorders.
However, they are not the only cause.
What other primary disorders cause sustained injury-thrombosis-inflammation,
thus leading to sustained acute inflammation with high-load exposure and then
immunization against normally safe or sequestered antigens? The list includes chronic or repetitive (1)
trauma and injury, (2) allergy-atopy and related granular leukocyte defenses,
(3) immunity and related lymphoid defenses, (4) infection-suppuration and
related neutrophil defenses, (5) thrombosis, i.e. anything that can
independently trigger, appropriately or inappropriately, acute reactive
inflammation or cause micro-thrombosis and necrosis. Note that this list implies sustained or
chronic states of these conditions.
Normal one-shot blood clotting after an injury or operation does not
induce wound and stromal auto-immunization.
Getting hives after eating a once-a-year strawberry will not cause
late problems, nor will getting a pimple or a cold or the occasional urinary
infection. Whatever the cause of acute
inflammation, it must be repetitive or sustained if the late phase
auto-immunization is to occur, such as we saw with hidradenitis. Of course, the whole story is a bit more
complicated. Acute infections can
induce this state, as can happen for rheumatic fever and Reiter’s
syndrome. Genetic variations also have
a role in some syndromes, such as HLA-B27 for ankylosing spondylitis and for
Reiter’s syndrome, HLA-B51 for Behçet’s syndrome, and MEFV for familial
Mediterranean fever. Regardless of the exceptions and complexities, the common thread
here is that under the right conditions that expose sequestered antigens,
auto-immunization can occur. What are
those “right conditions”? There is no
singular or dogmatic event that by itself is the unique cause of
auto-sensitization and the connective tissue disorders. If acute incidental chlamydia and
streptococcal infection can cause acute arthritis and acute carditis, it is
because those distinctive syndromes cause rapid exposure and sensitization of
occult auto-antigens. But acute onset
sensitization of this sort is the exception, not the rule. In most instances it would seem that
stromal auto-immunization and the cvd-ctd’s cook slowly and insidiously. With disorders of chronic primary
trauma-injury, allergy-atopy, immunity, infection, and thrombosis, the “right
conditions” are those of repetitive injury-thrombosis-inflammation leading to
sustained acute inflammation. In the
annals of medical research over the past 50-100 years, all of these factors (injury,
allergy, immunity, infection, thrombosis) have been implicated at some point
by some one as the cause of rheumatoid arthritis and similar diseases. None of them are “the” cause, but it is
easy to see how all of them are “a” cause, united by their ability to induce
sustained acute inflammation, which in turn leads to antigen exposure and
auto-sensitization, then followed by chronic auto-immune lymphoid
inflammation. It has been easy to demonstrate here the causal connection
between hypercoagulability and connective tissue disorders. Next consider the connection of cvd-ctd to
infection, a subject with considerable prior research. It has largely been speculative research
without definitive conclusion, but that is because such research wants to
find “THE” cause without appreciating that chronic infection can be just one
among several dynamically similar pathways to stromal auto-immunization and
the cvd-ctd’s. Nonetheless, such
research has implicated infections from all types of pathogens from a variety
of higher taxa – generally true pathogens not part of our healthy
human-bacterial eco-flora, including viruses, bacteria, mycoplasma, mycobacteria,
mycetes, and protozoa. Next, consider
the issue of chronic allergy. In
addition to the usual acute reaginic signs and symptoms that allergic
patients get, either incidentally, seasonally, or protracted from continuous
environmental exposure, many patients will eventually develop more diffuse
connective tissue symptoms. It is very
common for lupus and rheumatoid patients to have multiple allergies, often to
drugs but also to other natural and unnatural items. It is easy to dismiss this as a secondary
effect of their altered immune systems, a consequence of their cvd-ctd. Possibly, but it is also quite likely that
their heightened allergic sensitivity and sustained eosinophilic inflammation
is the primary state that eventually leads to auto-sensitization – cause
rather than effect of their cvd-ctd.
Among our hypercoagulable wound patients with auto-immune symptoms,
many start wound healing and also have improvement of systemic symptoms after
being anti-coagulated. So too, there
are many reports of putative “infectious rheumatoids” who have improved with
anti-microbial therapy. Treat the
primary cause of the sustained acute inflammation, and the secondary events
and clinical sequelae can then settle down. Once acute inflammation becomes prolonged and sustained, stromal
auto-immunization can then occur. On
slide 53 we will look more specifically at what happens after
auto-sensitization occurs, why the fibro-vascular stroma is so important in
this sensitization, and how this then leads to clinical connective tissue
disorders and impaired wound healing.
The purpose of this slide was to emphasize the quintessential central
role of sustained acute inflammation in this process, and to clarify that
sustained acute inflammation has several root causes in various primary
disorders that cause or induce acute injury, inflammation, and thrombosis. Caught in the middle of thrombosis and
inflammation are necrosis and ulceration, which when “chronic” and sustained
lead to the exposure of occult auto-antigens, eventually leading to
immunization against your own stromal cells and matrix. Remember what we said on
slide 12: “The importance of the gene
mutations must be emphasized. You
cannot cheat on a gene test, so when a patient has an altered gene and then a
bunch of other syndromic clinical problems, it is a good bet that the genetic
mutation is the root cause.” This is why so many hypercoagulable
patients have auto-immune disorders.
They are chronically making primary thrombi, causing necrosis,
inducing acute inflammation, and creating conditions that cause more
thrombosis, thus leading to the sustained state that eventually induces
immunity. The thrombotic condition
comes first, and the auto-immune state is an induced reaction. For these patients, their manifestations of
auto-immunity might be greater or lesser, an overt classifiable clinical
syndrome, or else a mixed set of autoimmune symptoms, or else just positive
laboratory serologies. Whatever the
severity and manifestations, when patients with factor V Leiden or
prothrombin 20210G genetic mutations have rip-roaring rheumatoid or lupus,
the cause-and-effect connection is real. |
|
|
|
|
|
This and the next 2 slides demonstrate the sustained primary
disease and inflammation and the cell admixture and lymphoid infiltration
that lead to wound auto-immunization and chronicity. The specimens on this slide are from a 74
year old woman with a history of thrombotic events and overt crippling
rheumatoid arthritis. She presented
with multifocal leg ulcers, some randomly placed, some associated with tendon
sheaths and synovitis. There were
distinctive thrombo-infarctive changes in some, inflammatory-lytic changes in
others. These longstanding
“refractory” ulcers gradually healed completely with anti-coagulation,
increased anti-inflammatory therapy, and skin reconstruction with
regenerative matrices. The images show vascular loci without significant acute
neutrophilic inflammation, nor are there any poly’s in the perivascular
connective matrix. However, the vascular
lumens are filled with acute and chronic thrombus including neutrophils. The vascular loci are hypertrophied due to
appropriate angiogenic activity and also to inappropriate infiltrates with
lymphocytes and plasmacytes. Something
about the vascular locus is attracting or harboring the lymphoid cells. The attraction to angiocytes and vascular
structures, especially those that are thrombosed would seem to imply some
sort of auto-sensitization. Of course,
this hypothesis cannot be proven by h&e histology alone. The lymphoid cells might just find the
vessels a homey place to hang out for reasons yet to be inferred or
discovered. However, given the
immunogenic and defensive role of these cells, and given the consistent
finding of lympho-plasmacytic infiltrates around vessels in certain classes
of impaired wounds, it is reasonable to assume that the lymphocytes are more
cause rather than effect of the wound impairments. |
|
|
|
|
|
These images are from a 33 year old man, who, after spine injury
and paraplegia, developed diffuse chronic pathological ulcers. The lesions all began with initial
blistering then persistent desquamation and failure to heal. The acute phase gross lesions and histology
were consistent with an eosinophilic or allergic process, bullous pemphigoid
being the “best fit” diagnosis. In the
chronic phases, the gross and histologic appearances were more inherently
chronic. The lesions improved with
steroids, but ultimately healed only with azathioprine therapy. (See slide 71 for gross images and more
history). The primary “pathogen” in
this situation was presumably some type of chemical exposure, e.g.
therapeutic pharmaceuticals prescribed during his post-trauma hospitalization
or else topicals related to wound care.
What could have, should have, would have been an easy problem to treat
if recognized and treated properly in the early phases became chronic and
refractory to any standard wound therapy.
Histology revealed the reasons why. Left: this view is below the wound surface in what
is nominally the angio-organization and histio-attraction strata of the
wound. There is edema mixed with
aminoglycans and scant if any collagen, yet there are large substantial
vessels, either only semi-organized, or else disordered by persistent angioid
activity and inflammatory infiltration.
(Impaired, disorganized, aberrant, and failed wound strata are common
findings in chronic and pathological wounds, especially autoimmune wounds.) The entire field is infiltrated with an
intense mix of “everything”, a CAP wound smorgasbord of all relevant cells
which contribute to its sustained impaired pathological state. There are numerous neutrophils and some
nuclear debris (in a healthy wound, poly’s would be nearly absent from this
stratum). There are monocytes-macrophages-histiocytes,
present in reasonable numbers, but in a deeper stratum where they would not
ordinarily appear. There are very
active angiocytes, some coalesced into vessels, others breaking away, many migrating
through the interstitial space, all morphologically typical of sustained
wound healing dynamics resulting from active growth factors affecting the
local load cells. The normal separation
of acute inflammation from wound module in time and space (see slide 52) has
failed, and these two populations are admixed, disrupting wound healing, and
exposing potential antigen processing cells to increased amounts of debris
and raw stromal detritus. Finally, the
wound, both vessels and interstitium, is overrun with lymphocytes, plasma
cells, and eosinophils. The
eosinophils are consistent with the presumed allergic-atopic pemphigoid
origin of the problem. The
lympho-plasmacytes reveal a latter stage of immune sensitization. In this case, the chronic allergic state is
probably the root or anchor of this system.
Chronic eosinophilic activity leads to sustained acute neutrophilic
inflammation. This in turn increases
the exposure of endo-cellular auto-antigens, as well as “stirring” the
architecture of the wound. Sooner or
later, there is enough admixture, in time and in space, of endogenous
antigen, antigen processing and presenting cells, and antigen receiving and
immunizing cells. Once the latter
phase of the process is reached, with antibody-bearing plasma cells, the
wound cannot heal. Knocking down the
chronic-immune inflammatory cohort by specific pharmaceuticals allowed the
wounds to heal. Right top: this is a
closer view of the activities.
Neutrophils, nuclear debris, monocytes and macrophages, angioid cells,
eosinophils, lymphocytes, and plasmacytes are all intermingled. Cells which do not belong in a normal
healthy one-shot wound are abundant, and even those cells that belong are
admixed abnormally. The very close
proximity and prolonged mutual contact of inflammatory, stromal, mononuclear,
and lymphoid cells is the “perfect storm” needed to transform normal healthy
wound refuse into stromal auto-immunization and wound disruption. Right
bottom: this is another close view
of a blood vessel in the wound. The
same abnormal pathological mix of cells is present. The angiocytes at face value are reasonably
normal and healthy. They have typical large nuclei and cytoplasm consistent
with a state of vegf and other growth factor induced hypertrophy and mitotic
and migratory activity. They are
partially but not tightly coalesced into a solid vessel. Migratory angiocytes at the periphery are
either landing and assembling onto the large vessel, or else lifting and
migrating away to areas of acute inflammation and mononuclear
chemotaxis. But all is not quite
right. For a vessel of this apparent
size, it lacks the cohesion and solidity expected. The interstitium of the vessel contains
leaked plasma (“fibrin cuffs”). Close
inspection shows that it is actually more plasma than angiocytes, a pattern
typical in immunopathic wounds of vessels in the (failed) angio-organization
layer. Infiltrated throughout the wall
of this proto-vessel are mononuclear cells and neutrophils. Surrounding the vessel are numerous plasma
cells. The failed organization of the
vessel, the targeted presence of immunogenic cells, and the sustentation of
acute inflammation are all prototypical of the auto-immune wound failure that
we are discussing. In preceding
examples we have seen, sustained microthrombosis was the primary cause of
auto-immunization and wound failure.
In this example, sustained allergy was the primary cause. |
|
|
|
|
|
Right: this is from an 84 year old woman with a
chronic radiation ulcer of the scalp.
We think we understand the pathophysiology of radiation wounds fairly
well. Photonic energy damages DNA and
nuclear machinery, by design. Cell
kill is latent, and becomes manifest during attempted mitosis. Highly proliferative dividing cells such as
cancers die quickly. Cells which have
no normal need to divide, such as mature stromal cells, can carry on
vegetative functions and survive.
However, when such cells are asked to divide, such as for wound
healing, the process dies with the cells.
Radiation injury is another example of where the intrinsic machinery
of healing is broken (although radiation wounds are just a tiny fraction of
all chronic wounds). However, a slide like this suggests that the problem may be more
complex. The histology throughout the
wound looked like a rock concert for plasma cells. Excessively laden with antibodies or golgi
apparatus, they exhibited every bizarre morphology of plasmacytes that
exists. Something in this chronic
wound has provoked the body to make immunoglobulins against itself. Radiation may have been the primary event
that caused the wound and invoked acute inflammation, but once the wound
became chronic, then something sensitized or immunized the patient to the
stroma. Perhaps radiation damaged
matrix chemicals, creating novel and immunogenic chemical species. Perhaps there was enough latent cell kill
that stromal cells degenerated at a sustained rate, increasing the load and
timewise exposure of endo-cellular debris in the face of persistent
ulceration and inflammation. (Remember
that since the leukocytes and lymphoid cells are marrow-derived and
blood-borne, they were not impaired by the radiation and could carry out
these acute and chronic reactive functions.)
The plasmacytes could of course be strictly non-cause-and-effect
coincidental to the wound healing impairments, but nonetheless, this is
another example of a particularly refractory wound having particularly heavy
infiltration with lymphoid “chronic inflammation”. Left: these two images come from the same wound,
a leg ulcer in a 31 year old woman with a primary coagulopathic disorder and
secondary immunopathy. She had a
typical history of thrombotic events along with miscellaneous immunopathic
symptoms. Laboratory evaluation
confirmed low protein C and low APC resistance (likely the primary problems),
a positive lupus anticoagulant (probably secondary due to induced
auto-immunity) and low factor IX (indirect evidence of a thrombotic state,
due to compensatory down-regulation of a prothrombotic element). The specimens are from the base and edge of
the ulcer, which grossly had intense vascular stasis, cyanosis, persistent
multifocal wound infarcts, wound pathergy (progressive infarcts with
debridement), persistent inflammation in spite of basic treatment, and failed
wound healing. Histologic features
(not necessarily seen in the two views selected) included: acute and chronic inflammation, acute and
chronic thrombosis, vascular necrosis or fibrinoid degeneration, vascular
disruption and disorganization, dense peri-vascular plasma cell infiltration,
interstitial necrosis, cellular debris and basophilia deeper than expected
for healthy wounds, cellular debris and basophilia along angiogenic cords,
scant or disorganized fibroplasia. Far left: A close view shows the angio-attraction
zone with numerous streaming angiocytes.
There is little evidence of angio-organization, in part because this
view may be too high or superficial in the wound, but in part because of the
diffuse admixture with plasma cells.
These cells presumably are attacking, degrading, or inhibiting
something in the local matrix or cell set, preventing proper wound module
dynamics and self-organization. Of
interest are the large macrophages with ingested red cells. Loose erythrocytes, i.e. “hemorrhage” are
normal and expected in this stratum, because organizing uncoalesced vessels
are still quite open and leaky. In
healthy wounds, a lot of this degrades and disappears. However, in some wounds macrophages
sequester the erythrocytes, degrade them within, and then they remain in situ,
seen grossly as long term hemosiderin staining in the skin. This is typical of venous ulcers and
various others, and it is not surprising here due to the intense vascular
stasis and congestion resulting from repetitive microthrombosis. Whether these hemosiderin laden histiocytes
are relevant to the issue of chronicity and auto-sensitization is unknown,
but it does reveal that macrophage activity is significant in this wound, and
may be part of the long term immune recognition and sensitization that has obviously
taken place, as evidenced by the numerous plasmacytes. Near
left: a wider view from a somewhat
deeper zone in the same wound.
Cylindrical vascular structures have formed, the largest one
conducting blood, but they are poorly organized, and overall expected wound
architecture is disorganized and imprecise.
The entire field is infiltrated with plasma cells and some
lymphocytes. One of the core issues of CAP and immunopathic wounds is that
latter phase impaired wound healing cannot be separated from acute phase
active injury. The ongoing active
injury is the chronic inflammation, triggered by autoimmunity against
cellular components (which was first created by prolonged acute inflammation
and necrosis due to an underlying primary disease, in this case a
hypercoagulopathy). Latter
inflammation and immunity are targeted against stromal elements, meaning ipso
facto that they are targeted against the wound healing machinery. Thus, in autoimmune wounds, impaired wound
healing and chronic inflammation are conjoined, with or without some acute
inflammation, with or without some degree of persistent thrombosis and
necrosis, and with varying degrees of impairment against the reparative wound
module and its elements. To the extent
that acute inflammation and thrombosis are continuously renewed, the whole
process becomes perpetuated and more entrenched. In the examples shown, it is easy to see histologically
this conjoined set of abnormal cells, all jumbled together, all mutually
doing things to each other that ultimately inhibit the wound. This patient did heal with a typical program of anticoagulation
and skin reconstruction with a regenerative matrix. However, prior to that it was refractory to
basic care. Persistent active
inflammation and necrosis in spite of treatment, along with failed wound
healing all meant that there was persistence of the pathological state, with
hypercoagulopathy being the primary “pathogen”. However, had hypercoagulability been the
only adverse factor, then anticoagulation alone (along with other basic care)
would have sufficed to heal the wound, as it does for many hypercoagulable
ulcers. In this case, something else
was also present sustaining and complexifying the wound and its responses to
treatment – chronic auto-immune lympho-plasmacytic inflammation. As can be seen in all of these cases,
untangling the interconnections of acute inflammation, chronic inflammation,
thrombosis, and their effects on subsequent wound healing becomes difficult, because this whole interactive
mess IS the intrinsic disease of wound healing. As we will see in Part 3, the physics of
complex systems explains why it cannot be so easily untangled. |
|
|
|
|
|
In the preceding few slides we looked at how primary
hypercoagulability and other primary causes of injury and inflammation lead
to sustained primary inflammation, a precursor to auto-immunization and
chronic lymphoid inflammation. We now
must show how these states explicitly cause stromal fibro-vascular auto-sensitization
which then leads to wound healing impairments and systemic connective tissue
disorders. To begin, here is a
reminder that wound repair is based on 2 cell types and their structures –
fibroblasts and angiocytes, and vessels and connective matrix. It is these elements which are caught in
the fracas of sustained inflammation, putting them in harms way of auto-sensitization. All injury and inflammation trigger repair. The mesenchymal component of normal wound
healing is the proliferative wound module of post-inflammatory repair. This depends on just two cells, histio-fibroblasts
& vascular angiocytes, which create the vascular and connective
structures which constitute the new stroma.
Top: three views of angiocytes and vessels. Left, loose angiogenic cords and
young streaming angiocytes in the aminoglycan angio-attraction layer. Center, early organized vessels
with still unsettled angiocytes, but conducting erythrocytes, at the
aminoglycan-collagen boundary. Right,
fully organized and maturing vessels in the deeper fibrous layers of the
wound. Middle: three views of
fibroblasts and connective matrix. Left,
young fibroblasts in loosely organized fibrillar matrix of the
histio-organization stratum. Center,
dense collagen trapping sequestered mature fibroblasts in deeper strata of
the wound. Right, maturing scar
with remodeling of fibers due to chronic maintenance functions of the
embedded fibroblasts. Bottom: three views of composite fibro-vascular
scar. Left, young scar, with
still loosely organized matrix and vessels, in the early healed wound. Center, fully proliferated scar,
dense with collagen and vessels, without fibrous interspaces or
compliance. Right, maturing scar,
regaining a histological architecture tending toward normal dermis or
fascia. Angiocytes and fibroblasts,
vessels and matrix – these are the only two cells and structures of the
generic stroma and of scar, which is nothing more than the stroma restoring
itself after injury. |
|
|
|
|
|
. . . and a final slide before showing how states of sustained
acute inflammation cause stromal fibro-vascular auto-sensitization with
subsequent wound healing and connective tissue disorders. This slide explains more thoroughly why
normal one-shot responses to injury do not cause the appearance and
recognition of stromal auto-antigens and subsequent sensitization. It is all related to the separation in time
and space of the various major events and cell populations which appear in
the integrated response of injury-inflammation-repair. Stromal auto-sensitization occurs when the
various events and populations, including the later abnormal “chronic”
lymphocytes all become stirred or mixed into the same soup or strata of the
wound. Left: a typical healthy open wound, a standard
vertical view showing the various time- and sequence-dependent strata of
normal healing. (see slide 6 and Part 1).
The strata are designated here as plasma protein and acute
inflammation (pp-ai), aminoglycan and angio-attraction (gag-attr), angiocyte
vertical migration zone (gag-vmig), the angio-organization and
histio-attraction layers (org), and then the organized fibroblast-connective
strata are grouped together (conn).
Adjacent to the photo and scale are four bars representing the
distribution and density of certain elements:
(1) acute inflammation (neutrophils, mononuclear leukocytes,
macrophages); (2) active angiocytes
(migrating and re-assembling, as opposed to fixed angiocytes in mature
vessels); (3) active fibroblasts
(organizing and making new matrix, as opposed to fixed fibroblasts trapped in
mature connective tissue); (4) time
(days, from 0 to 7). Recall that the
vertical anatomy of an open wound is also an archaeological dig into the
events of the past so many days. The
0-7 day scale shown is what would be happening in a normal healthy wound. Acute inflammation is the initial response to the injury. If it is a normal healthy one-shot
response, then it should tail-out by 3-4 days. We see this in the wound as neutrophils
which are dense at the top and then disappear within the angio-organization
layer. Active angiocytes appear in the
aminoglycan upper layers as migratory spindle cells and in early phases of
re-assembly. They become fully
organized in the angio-organization layer, where they also serve as the
mitotic source cells for new angiocytes that must migrate to higher
strata. Fibroblasts are starting to
appear at this level as well, in the early phases of migration and connective
proteogenesis. Below this level, in
the histio-organization layers, new vessels are mature and angiocytes are no
longer in an active phase. Fibroblasts
are at their peak of productive output and organization, and they too become
settled and mature in the strata below what is seen at the bottom of this
photo. Neutrophils are dense in the inflammatory layer. This is where injury, platelets and
thrombosis, and acute inflammatory signaling are all occurring. This is where host defense and protection
form the ambient world is occurring.
This is the plasma layer, the natural home of leukocytes. In the deeper parts of the aminoglycan
zone, where early vascular organization is starting, neutrophils are present,
but they are usually marginated in young vessels, and migrating out and
upward to where they are needed. This
is simply the disembarkation point for the neutrophil troops to start
marching to the front lines, but they have no inherent functions at this
level. Going deeper in a healthy
wound, neutrophils have no business – and no presence whatsoever – in the
underlying histio-attraction and organization layers. Thus, neutrophils and acute inflammation
have a moderate overlap with angiocytes in the upper aminoglycan strata, a weak
overlap with young vessels in the lower aminoglycan strata, and no overlap at
all with fibroblasts and connective matrix.
While monocyte-macrophages are far less numerous than neutrophils, as
acute inflammatory leukocytes the same patterns apply, including the limited
overlap patterns with the repair module cells underneath. Translated into time rather than spatial
coordinates, acute inflammatory leukocyte one-hot profiles peak at one day,
then tail-out around 3 days. Angiocyte migration is evident by 2-3 days, and early vessel
organization is present by 3-4 days.
Grossly, clinically, this correlates with the earliest appearance of
“granulation tissue” on a healthy wound surface as signs of inflammation are
subsiding (edema, erythema, pain).
Like everything else, angiocytes and vessels have their own one-shot
ramp-up-&-tail-out dynamics, subsiding (5-7 days) as active vascular
organization matures and new vessels become normal vessels with normal cell
sizes, geometries, metabolism, and blood conduction. The same is true for
histioblast-fibroblasts and the connective protein matrix, starting the
ramp-up at 5-6 days, and tailing out over 7-10 days or longer. The time estimates for these events are
approximate (depending on location on the body, age of the host, health and
co-morbidities, and assuming that the injury was a well-defined one-time
event). However, the inter-dynamics
between these populations, their scale-independent geometries and topologies,
they way they overlap with each other is true regardless of specific
times. Regarding angiocytes and
fibroblasts, they are both part of the proliferative repair module, they are
the two constituents of the generic stroma which is being restored, and they
are both the targets of auto-immune sensitization. The fact that they have their own
sequential one-shot cusps is of vital importance to normal wound-healing
physiology, but for the sake of understanding the origins chronic lymphoid
inflammatory pathology, they can be viewed as a coordinated unit. In summary, repair cells and structures (later, lower) normally
stay separated from acute inflammation cells and media (earlier, higher) by
time and by anatomical space. The
normal wound is a highly structured piece of anatomy that allows the defense
system to operate in the ‘war zone” at the top of the wound. The repair events take place in the
friendly territory behind the front lines.
The normal anatomy of a healthy wound ensures that the front line
defenses never really “see’ nor interact with the repair events behind. Obviously, if the defensive front lines
broke down, the body could be overwhelmed by xenobiotic invaders, so the
importance of the acute inflammatory front line cannot be overstated. However, it is also important that
neutrophils stay where they belong, because otherwise adverse events
happen. Neutrophils are inherently
defensive and destructive. Unlike
lymphocytes which have target-specific memory, neutrophil responses are
generic, preprogrammed, and heuristic, beating up anything that gets in their
way that they do not recognize. If
they are in the wrong strata, then they see things that they may be unfamiliar
with and the wrong (autogenous) things can get attacked. Furthermore, the activities of acute
inflammatory leukocytes can ultimately lead to the sensitization of
lymphocytes which eventually make and remember targeted antibodies against
specific antigens (providing for more defensive speed and accuracy the next
time that a specific enemy is encountered).
If acute inflammation is chewing up the wrong stuff, like angiocytes
and fibroblasts in the repair strata, then lymphoid sensitization against
autogenous elements is a risk. The
time and space separation of acute inflammation from repair, even within the
narrow bounds of just a few days and a millimeter or two, is a crucial aspect
of normal wound anatomy and physiology that preserves the health of the wound
and of the host. Problems happen when the strata and events of
injury-inflammation-repair get stirred and mixed up, allowing cells and
structures and activities which should be separated by time and space a
chance to mingle and intermix. When
non-specific neutrophilic defenses have a chance to “see” the repair elements
underneath, then the repair elements become just another target for the
neutrophils, eventually leading to lymphocytic auto-sensitization against
these elements. How is that the wound
can get jumbled and disorganized so that abnormal admixture can occur between
strata and cells? It happens when
there is some sort of abnormal re-triggering, feedback, and sustentation of
the wound via repetitive injury, thrombosis, or inflammation. These dynamics were explained on slides 46
& 47. The implication there was
that disorder and chaos are timewise dynamics, with the wound phases all
becoming concurrent and simultaneous.
However, as this timewise confusion of events unravels, the same thing
is happening to the spatial organization of the wound, with the various
well-defined strata losing their individual identities as inflammatory and
repair cells, structures, and matrix start to intermix. The general effect is twofold. First, there is a general inhibition of
repair events as defensive leukocytes and acute inflammatory chemistry
impede, damage, suppress, or inhibit the ability of repair cells to
function. Second, the systems that
allow acute inflammation to identify and process antigens for the sake of
immunity – “memorizing” the defense – can now be directed against autogenous
repair elements that are normally not seen by the defenses, leading to
stromal auto-immunization which further disrupts the activities of the repair
system. Right: a leg ulcer in a patient with polyarteritis
nodosa. The inflammatory infiltrates
are largely neutrophils, although plasma cells and lymphocytes are also
present. However, rather than being at
the top of the wound where they belong, the neutrophils are present
throughout, especially concentrated around vessels. There are no old blood vessels in this area
(other than the sclerotic one at bottom right), just regenerative new vessels
in what would otherwise be angio-organization strata. Attempted angiogenesis is mixed with
intense acute inflammation attacking those new vascular loci. The diffuse distribution of the neutrophils
means that new wound then new angiogenesis then new inflammation is occurring
or being perpetuated everywhere.
Remnants of normal dermis are at the bottom, but above that there is
little organization or distinction of different structures or strata, with
acute inflammation extending deeply into the whole process. Center: an ulcer that occurred spontaneously in a
previously healed skin grafts of the abdomen.
The history is on slide 57, where you can also see the active
dissolution of the epidermis at the margin of the ulcer caused by acute
neutrophil activity triggered by dense lympho-plasmacytic infiltrates within
a vascular locus. On this image, focus
on the mixing of the wound. The normal
strata are disrupted. There is a
slight top layer consistent with the normal plasma protein and acute
inflammation stratum, and at the bottom there is a more normal young fibrous
matrix (horizontally oriented zone).
However, most of this view in between is rather homogeneous where
there should be defined stratified anatomy.
Aminoglycan strata are hard to discriminate from plasma zones and
young fibrous zones. Streaming
migratory angiocytes are abundant, but the vertical migration zone is gone,
and established “granulation tissue” types of vessels are present at all
levels, right to the very surface.
Neutrophils are everywhere, at all levels, and so too are mononuclear
leukocytes (in correspondingly much smaller numbers), but so too are a rich
infiltrate of lymphocytes, plasma cells, and eosinophils. Reflecting this indiscriminate cell
admixture, there is destruction, inhibition, and disorganization everywhere,
with no respect to the separation or stratification of time and anatomy that
characterize normal wound histology and physiology. |
|
|
|
|
|
The last several slides, 46 – 52, focused on the nature and
causes of sustained inflammation.
Repetitive injury, thrombosis, and various forms of acute inflammation
all lead to sustentation and self-perpetuation of the whole process,
eventually causing abnormal admixture of inflammation and repair populations
in time and space. We have already discussed
that this eventually causes lymphoid auto-sensitization. On this slide, we will look more closely at
the consequences of sustained inflammation and how that leads to
auto-immunization, specifically how auto-immunity results against stromal and
connective tissue elements, including the wound. The text of this slide is simply a reprise of slide 40. The main points made were: (1) Auto-sensitization depends on recognition,
processing, & presentation of auto-antigens to lymphocytes. (2) One
general way this happens is by exposure of occult & sequestered antigens. As a consequence of acute inflammation,
endocellular and endonuclear debris is released which can lead to the
formation of intra-cytoplasmic and anti-nuclear antibodies. Some of these are generic anti-cellular
antigens, but some will be directed against tissue-specific antigens, as
occurs with hidradenitis and uveitis.
Some can also cause antigen cross-reactivity, such as seen with latex
& spina bifida. (3) Another general way that auto-immunity
occurs is by sensitization against normally open and exposed non-antigenic
chemicals that become modified by opsonization or (inverse) haptenization,
such as occurs with rheumatic carditis & parf and with Reiter’s syndrome. (4)
Once auto-antigens are created or exposed by acute inflammation, they
are processed by monocyte-macrophage-histiocytes and then presented to
chronic lympho-plasmacytic immune cells.
The creation and exposure of cellular or matrix auto-antigens is
unlikely and irrelevant during normal physiological one-shot injury-thrombosis-inflammation-repair. However, these events become ever more
likely as inflammation is sustained, eventually inducing a state of chronic
auto-immune lymphoid inflammation. Focus now on the issue of cell and
tissue specific debris and antigens. The
connective tissue disorders have a variety of distinctive “selective target”
sensitizations, such as uveitis, urethritis, and sialoadenitis. Tissue specific antibodies are crucial to
the syndromic expression of many auto-immune diseases. For example, the “rheumatoid factor” is a
common auto-antibody seen in many auto-immune states, but the anti-ccp is
considered quite specific for rheumatoid.
What distinguishes rheumatoid from other cvd-ctd is its primary target
and effects against synovium, so there is something quite specific about the
target tissue and the distinctive antibody.
These concepts can be further illustrated via hidradenitis
suppurativa, an acne involving apocrine glands. Like any acne, early disease starts with
ductal hyperplasia and obstruction causing micro-abscesses. However, chronic active disease is marked
by lymphoid infiltrates around the glands, an auto-immune component of the
disease that is recognized as having a role in perpetuating the apocrinitis. It should not be surprising that
auto-immunity occurs, since the micro-abscesses and glandular “rupture” will
expose sebaceous and other epithelial antigens repetitively and chronically
to the mesenchyme. This is just like
hypercoagulability in that it creates the perfect chronic mix of sustained
acute inflammation and macrophages, novel or occult antigens, and then
lymphocytes and chronic inflammation.
For hidradenitis, sensitization is against the glandular epithelium,
leading to chronic apocrinitis and persistence of the acneform state. For hypercoagulable states, sensitization
occurs against vessels, leading to further arteritis. What is interesting though is that once
auto-immunization occurs, it can have global effects. The patient shown on slide 40 had active
untreated hidradenitis for over thirty years.
Excision of the affected areas cured the problem. In so doing, many other symptoms that she
had lived with for years suddenly resolved.
Along with general malaise, she had symmetrical large joint
polyarthralgias, sore wrists and hands, morning stiffness, and sicca
syndrome. All of this evaporated as
the post-excisional wounds healed.
This story is not unique, neither among hidradenitis patients nor
among any patient with a chronic inflammatory state. Since our interest here is on
the wound and wound repair, consider next the mechanisms whereby
auto-sensitization can occur to wound specific structures and debris. After any injury, the generic response
is the inflammation and wound healing process. Wound healing is simply the generic stroma
restoring itself for the sake of restoring structural integrity of the
body. Multicellular life, with its
division of labor between specialized cells, tissues, and organs, has two
quintessential requirements for existence.
(Unicellular species worked out the basic issues of life itself; we are talking here about what is then
required for multiple cells to assemble into a composite lifeform.) First, multicellular life needs a
structural framework that cells can hang on to. Second, the organism needs a distribution
system that allows all cells to send and receive biochemicals to each other
for the sake of communication, functional integration, and metabolic
sharing. Even in a species as primitive
as a jellyfish, ectodermal cells in the tentacles are tasked with capturing
food, entodermal cells in the gastric pouch are tasked with digesting food,
and muscle cells in the mesoglea are tasked with moving the organism here and
there to find food. There must be a
way for the muscle and ectodermal cells to get their fair share of nutrients
from the digestive cells. The vascular
extensions of the gastrovascular cavity are the distribution pathways that
allow the digestate to circulate.
These are the evolutionary precursors of our own blood vessels. Regardless of how complex multicellular
life has become in the higher chordates, it all rests on two mandatory
underpinnings of multicellular life – a framework to allow it all to cling
together, and a vascular distribution system to allow specialized cells to
share substrate and metabolic products.
In animals, the structural framework that ascended was based on
collagen and connective proteins. The
standard vascular distribution system that developed was a space-driven
fractal r-net of regulated density governed by closed loop control between
parenchymal cells and the vascular cells – i.e. our blood vessels. (There is more about this in the epilogue
after slide 59.) Without a structural
framework and a vascular distribution system, complex self-contained
multicellular life as we understand it could not exist. The generic stroma is nothing more than
this basic fibro-vascular infrastructure that allows multicellular organisms
to hang together. It is the framing
and utilities of a new building, the chassis and bus wires of an electronic
machine. Everything else that makes
that building a home or office, that chassis a computer or television, or
that stroma a bivalve mollusk or a bipedal mammal occurs because specific
parenchymal elements get pinned to that framework. When an injury occurs, the response of the
host is to create new stroma to hold everything back together. That is what wound healing is, just the
reformation and restructuring a generic stroma. It is based on two, and only two, general
elements – fibroblasts which make the structural matrix, and angiocytes which
make the vascular distribution system. We have made the case that under
conditions of sustained acute inflammation, that pathological conditions are
created. The wound gets stirred, cell
populations get intermixed, and the load of inflammatory debris goes up,
including the exposure or creation of potential auto-antigens. This brings us to the question, what
auto-antigens? What is present in the
stew of sustained inflammation that can be revealed or altered enough that
the body’s defenses will perceive it as unnatural and worthy of immunization? In principle, a wide variety of
chemicals could be the antigens, coming from any and all tissues, depending
on where, why, and other circumstances of the sustained inflammation. However, our focus here is on the generic injury-infarct-wound. What is in the sustained generic
wound? Acute inflammation interspersed
with the stroma itself. Acute
inflammation leaves behind its own degradative debris (leukocytic “poly
dust”). It also leaves behind
degradation debris of the generic stroma – angiocytes, fibroblasts, vessels,
and matrix – that have been damaged and renewed, damaged and renewed. Also in the mix are the active and alive
fibro-vascular structures and cells of the stroma. The antigens being released or exposed in
the sustained acute wound are in, on, and derived from the stroma. The debris that macrophages are cleaning up
are the degraded stromal elements and antigens. When lymphocytes start to appear and
macrophages have opportunities to present antigens to them, it is stromal
degradation products which are exchanged.
Whether these are occult endocellular chemicals or else open matrix
chemicals which have been modified-opsonized-haptenized, the net effect is
the same. Once lymphocytes and plasma
cells have become immunized against one of these antigens, you are now
immunized against your own stroma. And
the stroma is not unique to the wound.
On the contrary, the wound is just a specific instance of “stroma”,
repairing itself after disruption.
Thus auto-immunization against stroma means negative effects on the
wound, but it also means effects on the general stroma throughout the body,
thus becoming the collagen-vascular diseases and connective tissue disorders. At this point you might ask, “what if
there is no wound? . . . what if the patient just has some minor trauma or
synovitis or a little blood clot with a minor microscopic infarct? . . . do
the same events happen if there is no real wound?” The answer is twofold. First, we are not talking about incidental
events – they are safe from auto-sensitization. Only sustained conditions have this
risk. The second answer is that these
scenarios are all wounds. Any little
thrombus and infarct will trigger inflammation or necrosis. Any allergy-atopy event will trigger acute
inflammation. Any flare-up of a
granulomatous or atypical infection, any traumatic bump or ding, any
inflammatory event due to any primary event – all – all will result in the
cycle of injury recognition then thrombosis then acute inflammation then
repair. Each and everyone of these
events, by virtue of the fact that they trigger the inflammation-then-repair
process is ipso facto a wound. And the
key to stromal sensitization is in that fact, that repair events are
triggered, meaning that angiocytes and fibroblasts must show up. MUST.
Any primary thrombosis, infarct, injury, inflammation, allergy, immune
event, etc. – any and all trigger the sequence of events that leads to the
repair phase, and angiocytes and fibroblasts must be invited. Their presence is unavoidable and
inescapable. So, when the wound
becomes sustained, and cells and structures and strata are mixed, and
phagocytes are processing lots of debris and trying to feed it to immune
lymphoid cells that are appearing, then angiocytes and fibroblasts are what
is caught in the melee. If the
sustained problem is exposing potential parenchymal antigens because the
process is in the liver or thyroid or stomach, then sensitization to specific
organs might occur (and there are certainly many diseases of parenchymal
auto-immunity). But regardless of
where the problem is, anything that triggers injury-infarct-inflammation must
perforce trigger angiocytes and fibroblasts, vessels and connective matrix,
and thus these cells and structures will ALWAYS be in the center of those
activities which induce tissue auto-immunization. Thus, stromal auto-immunization is almost
inescapable under the right circumstances of sustentation or chronicity,
which is why wound healing problems and connective tissue diseases are not
such rare or unusual disorders. |
|
|
|
|
|
Summary of
sustained acute inflammation and the induction of stromal auto-immunization We have been looking at the chain of events starting with a
primary state of injury-thrombosis-inflammation, then sustained acute
inflammation, then stromal auto-sensitization, then clinical sequelae of
auto-immunization. This process does
not happen with normal one-shot injury and acute inflammation, because debris
loads are smaller and cell populations do not get mixed. This pathology occurs when there is
sustained acute inflammation, which can result from any number of underlying
primary disorders. This sustained state
exposes various sequestered endocellular chemicals or creates novel chemicals
from open matrix, leading to autoimmunity which occurs when the immune system
“sees” these antigens. The generic
stroma is not only caught in the middle of this sustained inflammation and
sensitization, but the presence of angiocytes, fibroblasts, and their
derivative structures is an inevitable consequence of any of the primary
wound inducers (injury-thrombosis, inflammation). The obligatory presence these stromal
elements means that angiocytes and fibroblasts, vessels and matrix are in
harm’s way regarding immune recognition, eventually leading to stromal
auto-immunization. This pathology also lets us answer the question what came first,
thrombosis or inflammation, hypercoagulability or autoimmunity, the
thrombophilic disorder or the connective tissue disorder? The thrombotic disorder is the cause – or
the chronic primary infection, or the allergic-atopic state or the repetitive
trauma – they all come first, and the connective tissue disorder is a
consequence of the sustained state of injury-thrombosis-acute inflammation
that then leads to lymphoid auto-immunization. The more severe and more sustained the
primary disorder, the greater the chances of auto-sensitization. That is why so many hypercoagulable
patients have auto-immune disorders, be it an overt classifiable clinical
syndrome, or else a mixed set of autoimmune symptoms, or else just positive
laboratory serologies. When patients
with factor V Leiden or prothrombin 20210G genetic mutations have rip-roaring
rheumatoid or lupus, the cause-and-effect connection is real. Note that the lymphocytes and plasma cells which are the agents
of auto-immunity are not inherently disordered or diseased. They are behaving properly in response to
the conditions that they are presented with (more on this in Part 3). The whole immune system is behaving the way
it is meant to, but it gets confused about who the enemy is when primary
acute inflammation is sustained, rather than being a one-shot, and wound
structures and phases then become corrupted and intermixed. Once stromal auto-immunization occurs, with
vessels and connective matrix as the targets, then protean clinical sequelae
can ensue. Chronic lympho-plasmacytic
inflammation can appear in the wound, disrupting its functions. Systemic effects and disorders of the
generalized stroma lead to the connective tissue collagen-vascular disorders, On the following few slides, we will look
specifically at how wound healing is altered in the presence of the
autoimmune connective diseases, and then make the formal connection between
autoimmunopathy, connective tissue disorders, and altered wound healing. |
|
|
|
|
|
In the last several slides (46 – 53), we looked at how the
intermix of acute inflammation, sustained acute inflammation, and chronic
inflammation occurs, induced by some sort of sustained primary disease or
injury that creates repetitive inflammation, thrombosis, necrosis, or allergy
and immunity. We looked at the
histopathology of that cell mix in chronic non-healing pathological wounds,
and why that cell mix is at variance with the normal spatial and temporal
distribution of cells in a wound, We
then addressed the consequences of that cell intermix, leading to stromal
auto-immunization. Since the wound is
nothing more than the fibro-vascular stroma putting itself back together
after injury, pathologies of the stroma should affect wound healing, and that
is precisely what happens. In addition
to their clinical and dynamical effects, immunopathic and other chronic and
pathological ulcers have distinctive histological variances from normal wound
healing (in addition to signs of the primary pathology). Seen here is a sampler of effects. Other examples follow on the next few slides,
followed by a more comprehensive listing of observable changes in
immunopathic wounds. Complex
inflammation profiles: shown is a
chronic non-healing popliteal ulcer, free of significant gross inflammation,
but with diffuse neutrophil infiltrates in the tissues and packed in
organizing vessels. Also visible is an
angiocyte mitosis, which were numerous, as many as might be seen in some
cancers. Lympho-plasmacytic infiltrates:
these should be familiar by now, after looking at the examples on
preceding slides. Lymphocytes have
little presence in a normal healthy one-shot wound, and plasmacytes
essentially none. Dense infiltrates
are of pathological significance and correlate with wound
refractoriness. Acute-chronic population admixture: likewise, we have just looked at
examples. It is the intermix of the
acute (neutrophilic) and chronic (lymphocytic) cell populations which assures
perpetuation of the wound. Failed and missing wound strata: in the sample shown of a particularly long
standing and refractory ulcer, the inflammatory and plasma strata at the top
give way to a fibrous histio-organization layer without any overt
aminoglycans or mid-strata of the wound.
Bizarre cell kinetics and
disorganization: an organizing
vascular locus is seen with disoriented misdirected angiocytes and a plasma
protein cuff that sits inside rather than outside. Epithelial
arrest: a hallmark of many
non-healing wounds, this is one feature that is easy to observe grossly. The acute abruption of the epidermis, without
any migration or squamous metaplasia, is a distinctive feature of arrested
wounds. |
|
|
|
|
|
This and the next 3 slides will illustrate aberrant wound
histology in patients with a priori connective tissue diseases, underlying
causative disorders, or localized wound auto-immunization under conditions of
chronicity and sustained injury. Some
of these examples illustrate acute destructive events “caught in the act” of
active ulceration, whereas others are samples of chronic non-healing wounds
illustrating wound healing impairments from the chronic and admixed
inflammation. Shown here is an abdominal wound in a 53 year old woman with
lupus since age 18. The patient
minimized her lupus history and symptoms, and she was on no active
treatment. However, she had a variety
of features such as a malar rash, sicca, arthralgias, malaise, etc. She was also chronically short of breath
from an undiagnosed non-emphysematous cystic degenerative lung disorder which
also proved to be due to lupus. A week
or two after otherwise uncomplicated abdominal surgery, she started to
develop active necrosis of the scars and wound margins. The active ulceration was of the
inflammatory-lytic pattern. Biopsy, as
shown here, confirmed that this was a lupus related event. With regard to wound pathologies, the dissolution
of new and old scars is a not-too-unusual distinctive complication of systemic
lupus erythmatosus. She was started on
steroids, and all of her symptoms improved, and the wounds stopped dissolving
and then healed after a few weeks of basic topical care. Left: this view comes from the skin at the margin
of active dermal lysis and ulceration.
It shows a vascular locus within the dermis. The dermal architecture was normal and free
of acute inflammation. However, most
vessels in the dermis had infiltration with lymphocytes and plasma
cells. The lymphoid infiltration
became dramatically intense, as seen here, at the margin of ulceration. In this view, large pale angiocytes look
intrinsically normal, but are somewhat disorganized among the
lymphocytes. The infiltrate is
predominantly lymphocytes. Right: seen here is the nearby zone of active
lysis and ulceration. Note that this
pathology was acute but not sudden (sudden, as might happen with a vascular
infarct or suppurative abscess). The
“slow burn” of lysis over a period of two weeks allowed wound healing events
such as angiocyte proliferation and migration to occur. Thus this view does show migratory angiocytes
and early vascular structures.
Intermixed are neutrophils, poly dust, monocyte-macrophages,
eosinophils, lymphocytes, and plasma cells.
It is another “perfect storm” of reactive destructive inflammation,
wound detritus, opportunities to process antigens and trigger sensitization,
and then have chronic immunized cells retrigger these events, destroying
tissue and disrupting healing. |
|
|
|
|
|
Left: is an 81 year old woman with a chronic refractory
ankle ulcer following radiation for melanoma.
The tibialis anterior tendon was also open and shearing in the
wound. Skin reconstruction was done
with a regenerative matrix. Rapid
total re-ulceration occurred unexpectedly, without any seeming
provocation. This cycle and events
repeated, always with a typical inflammatory-lytic pattern of
ulceration. The images are from a
biopsy at the edge of active ulceration during the latest rapid
re-ulceration. There was typical
necrosis and non-specific acute inflammation in the ulcer itself. However, the dramatic findings were the
lymphoid infiltrates, occurring in the ulcer and in the skin near the wound margins,
as seen here. Far left: essentially
every vessel and vascular locus in the scar or dermis at the margins had an
intense infiltration with lymphoid cells, mostly plasmacytes. These infiltrates lessened and disappeared
at deeper layers, a few millimeters down, but near the epidermis and wound
surface, virtually no vessel was spared.
Near left top: is a close view of the infiltrates, mostly
plasma cells densely packed around blood vessels. Near
left bottom: is another close
view, near the edge of active ulceration.
The vessels traversing the lymphoid aggregates are laden with
neutrophils. This was an unusually
difficult ulcer to heal and keep healed,
This is another case (see slide 50-right) where there was more to the
wound impairment than just the nominal history of radiation. One can speculate on the origins of the
lymphoid sensitization in this case, but radiation injury, sustained acute
inflammation, and prolonged time probably all had a role. Right: is a 65 year old woman with a chronic
refractory leg ulcer. History and
laboratory profile were consistent with a hypercoagulable state, and she had
active rheumatoid arthritis. Just as
for the radiation case on the left, neutrophils in this specimen were
generally sparse and consistent with generic acute inflammation (as opposed
to the intense infiltration that would be seen with suppuration,
polyarteritis, neutrophilic dermatoses, etc).
The prominent pathology is the lymphoid infiltrates around vessels and
within the vascular loci. All three of
the images demonstrate the infiltrates and aggregates, made predominantly of
lymphocytes with some plasma cells.
This ulcer also went through cycles of healing then re-infarction and
re-ulceration over a year before finally healing and remaining stable. |
|
|
|
|
|
This is a 55 year old woman who had a chronic
abdominal wound over a large ventral hernia.
Her history was otherwise benign.
The plan was to get the open abdominal wound healed, let it mature for
so many months, then do a definitive abdominal wall reconstruction. Ultimately that plan was successfully
implemented with good final results.
However, prior to reconstruction she went through a period in which
the long-healed regenerative epithelium on the old wound started to
spontaneously re-ulcerate for no overt reason or provocation. Biopsy showed the findings illustrated
here. Steroids helped control the
ulceration, and later allowed the hernia to be fixed without incident. Left: is a wide view of the edge of one of the
actively enlarging lesions. The open
ulcer is at the left The dermis or
scar is inflamed in the upper strata, with a mix of acute and chronic
inflammatory cells. At center is the
edge of the epidermis, and to the right the epithelium sits over “granulation
tissue” of excess hyperemic vessels.
In the center and at the far upper right, note the presence of the
basal layer (stratum germinativum) and the relative thickness of the
epidermis. In between, the epidermis
is thinner, with loss of the basal layer.
Below this is an area of basophilia and darker cells interspersed with
the blood vessels. Right upper & right lower: are progressively zoomed in views of this
area of interest. The lower layers of
the epidermis have been eroded by a front of acute leukocytic inflammation,
predominated by neutrophils, but with a fair number of eosinophils. This acute inflammation sits above a zone
of lymphoid infiltrates, recognized by their round dark nuclei. These infiltrates have plasma cells but
mostly lymphocytes. They are
interspersed within a vascular locus of regenerated immature vessels and
migratory angiocytes. It would appear
that the chronic lymphoid inflammation triggered an acute neutrophilic event
which is the immediate cause of the ulcer.
Why the lymphoid cells are attracted to the vascular locus presumably
represents sensitization that developed in the past during prolonged periods
of acute inflammation and persistent ulceration. |
|
|
|
|
|
This is a 65 year old woman with a 10 year history of
scleroderma-crest with other rheumatoid or mixed connective tissue symptoms,
presenting with a chronic ulcer at mid leg over the tibia. The patient has a history of
thrombophlebitis and miscarriage, and she is Factor V Leiden heterozygous. The gross findings show infarction as well
as lysis, and histology of the excised wound shows acute and chronic thrombi
and peri-thrombotic infarcts. Right: the gross appearance of the ulcer at the
time of consultation. The erythema and
dermal lysis at the 6 through 9 o’clock position is a typical
inflammatory-lytic pattern of ulceration, whereas the yellow fat necrosis at
the base and the black shreds of dermis are characteristic of
thrombo-infarction. Center left: is from an area of necrosis. There is thrombus and organization in
vessels. Lymphoid cells are present,
but they are few compared to the dense neutrophils. The darker more homogeneous basophilic
material is necrosis, as expected around the thrombosed vessels, Left: is from the wound in an area of
viability. There is no necrosis here. Scar or dermis (pink areas) is relatively
bland, without diffuse or concentrated neutrophilic infiltrates. The basophilic nodule in the center is due
to dense cellularity. This is a
vascular locus with typical hypertrophic and migratory angiocytes, dense
vessels, and also some fibroblasts and young matrix, i.e. relatively normal
“granulation tissue”. However, it is
heavily surrounded and infiltrated with lymphoid cells, mostly lymphocytes
with some plasma cells. While
neutrophils are not especially prominent in the pink matrix areas, they are
abundant within the lymphocyte-infiltrated central vessels. Center
right: another view under the
ulcerated surface. In the pink
collagen matrix areas of the dermis or fascias, mixed acute and chronic
inflammation are present, but inflammatory cell density is fairly low. (The wound had been cared for with good
hygiene and topical silver sulfadiazine, so the inflammatory layer at the top
is sparse and there is no suppuration.)
The dominant finding is the intense lymphoid infiltrates surrounding. Something has attracted immune cells to the
vessels. The thesis presented here is
that it is the primary hypercoagulable state which has caused repetitive
opportunities for immune cells to recognize components of the vascular
locus. Once the system is
auto-sensitized, some influence or activity of the lymphoid cells triggers
neutrophil attraction and activation, leading to ulceration, and then
interfering with healing. |
|
|
|
|
|
Here is a list of common histological features and variances
from normality that can be seen in chronic and pathological wounds that are
due to auto-immunity, collagen-vascular disease, and chronic lymphoid
inflammation. These are changes
observable with routine light microscopy and stains such as hematoxylin-eosin
or trichrome. Even more features are
revealed by immuno-staining. Active disease
and injury: These include signs of
the primary underlying disease or injury that led to the state of sustained
acute inflammation and then lymphoid immunity. These also include signs of active injury
and ulceration. For coagulopathic
states and thrombo-infarctive patterns of injury, what can be observed
includes thrombosis, vascular necrosis, vascular hyalinization, and tissue necrosis
and wound infarcts. For acute phase
inflammatory-lytic ulceration, active neutrophilic epidermolysis can be seen,
along with generalized neutrophilic and mononuclear inflammation within
dermis or other mesenchyme. Acute inflammation: Acute inflammation is present to some
greater or lesser extent in virtually all wounds and injuries. (Since the subject here is auto-immune
wounds, this discussion excludes abscesses and suppuration and similar
pathologies where extensive and aggressive neutrophilic inflammation is
supposed to be there.) What is implied
here is not the normal incidental one-shot reactivity to the primary injury
that exists in the upper wound strata, but rather the sustained acute
inflammation that results from some prolonged injury, pathology, or other
perpetuation of the process. It also
implies the spatial as well as timewise scrambling of the process, with
admixture into other strata or cell domains where it should not normally be. These findings include acute or sustained primary
neutrophilic inflammation not due to injury or other overt exogenous provocation.
Acute inflammation can be present in
deeper wound strata (the various fibroblast and connective matrix strata)
where neutrophils normally should not be present (this is the spatial mixing
that can lead to eventual stromal auto-immunization). Acute inflammation (neutrophilic and
mononuclear) can be intermixed with chronic inflammation (lymphocytes and
plasmacytes), a distinctive aberration from normal wound healing. Acute inflammation can be targeted against
various stromal structures or strata, such as seen in non-specific
leukocytoclastic vasculitis, polyarteritis and similar immunopathic
arteritides, and immunopathic dermatoses and panniculopathies. Coagulative lysis or dissolution of tissue
(as opposed to infarctive necrosis) will be present in areas of neutrophil
concentration or activation.
Eosinophils, as mediators of allergy, may be scattered
non-specifically in inflamed areas (like the normal non-specific presence of
neutrophils), but they can also be seen admixed with neutrophils in the dense
forward edge of active tissue lysis. Chronic
inflammation: Pathologists describe
“chronic inflammation” as lymphocytes, plasma cells, and eosinophils. As we are starting to see here, and as will
be presented in detail in Part 3, the presence of these cells has serious
implications and effects related to immunity and auto-immunity. “Chronicity” in the sense of “long
duration” is not the biological implications of these cells. Nonetheless, they are late phase
constituents which mark the onset of chronicity and intrinsic pathology in
the wound, and they do not belong in a normal healthy one-shot wound. Observable changes include lymphocyte
infiltrates, plasma cell infiltrates, and eosinophil infiltrates. There may be mixed “chronic inflammation” with
admixture of the three cell types, but oftentimes one cell type will
predominate. Chronic vascular or
perivascular infiltrates and aggregates of these cells, especially
lymphocytes and plasmacytes, are a distinctive feature of the most
“intrinsified” and refractory ulcers.
Diffuse interstitial lymphoplasmacytic “chronic inflammation” is seen
in wounds undergoing active lysis and dissolution, and in those which are
“stable” but refractory and persistent. Admixture: The significance of admixture was explained
on slide 52 (and others). During
normal incidental one-shot reactive wound healing, the acute inflammation
module (AI) and the wound healing module (WM) are contingent events with
dependent blocks on the control loop, but they remain largely separated in time (wound phases) and in space (wound
strata). Under chronic and
pathological circumstances, that separation breaks down, and it is the admixture
which is a crucial component of auto-immunization, chronic inflammation (CI),
and “intrinsification” of the wound.
Histologically, this is seen as complex inflammation profiles, in
which acute inflammatory cells are present in many strata, side-by-side with
chronic and immune inflammatory cells.
Monocytes (AI) mix with lymphocytes (CI), an opportunity for antigen
presentation. Neutrophils (AI) are
interspersed with blood vessels and fibroblasts (WM), an opportunity for
lysis and disorganization of repair elements.
A dispersed “well-stirred” intermix of acute-chronic inflammation can
be seen in two places: (1) the
developing ulcer, i.e. the zone of active acute necrosis and lysis, and (2)
the chronic established wound where gross active necrosis is controlled, but
the wound is not healing. In areas of
active ulceration, neutrophil activation and tissue lysis can also be seen
adjacent to chronic lymphoid aggregates, presumably being triggered to
activity by the lymphocytes. Disordered
proliferation: Whatever the detailed
biological reasons why the presence of chronic lymphocytic inflammation
inhibits or disrupts wound healing, those disruptive effects can be seen in
the failing wound. What can be seen
are (1) disorders of specific cells and structures, and (2) failure of
multiple cells and structures to organize and assemble. Regarding the appearance individual
elements, what can be seen are bizarre cell kinetics (mitosis and evidence of
proliferation, without cell persistence and tissue formation), cell
disorganization (failure of cells to appear in the correct strata or with
proper density or census), vascular disorganization (failure of angiocytes to
coalesce into proper vessels and patterns), and fibrous disorganization
(failure of fibroblasts to appear where expected). Epithelial growth can be completely
arrested. Failed
organization: Failure of wound
structures to assemble properly is manifest as a changes in the overall
architecture or histo-anatomy of the wound.
This includes “blurring”, “diffusion”, overlapping, and other
intermixing or loss of definition between normal wound strata. Certain strata may be failed, unusually
thin, or missing altogether. Loss of the
aminoglycan layer is common. Since
this layer is essential for normal vascular attraction and assembly, the
vertical migration zone (angio-attraction and angio-organization strata) is
also disrupted. Variations or absence
of the other proliferative zones (histio-attraction and organization) also
occurs. It is interesting that many
chronic wounds do become progressively fibrotic as time goes on, due to slow
fibroblast activity and fibroplasia in established deep strata at the base of
the wound. However, they refuse to
epithelialize, and absence of the aminoglycan and proliferative strata, aka
“granulation tissue” is key to epithelial arrest. In other wounds with hypertrophic
granulation tissue, these strata can be unusually thick, yet angiogenesis and
fibroplasia may be disorganized.
Variations in strata means that their relative thickness and
“anatomical depth” of the wound can be altered, and so too the absolute
physical depth, either greater or lesser than normal or expected. These aberrant histological changes are to be expected in people
with CVD-CTD. However, they also in wounds
and patients with immunopathic symptoms (without a strict nosological
“rheumatological” diagnosis). They
also occur in those patients with underlying primary risk factors (e.g.
hypercoagulability and allergic-atopic states), and even in those with “no
history” where wound appearance and behavior are altered in ways consistent
with these pathological states. |
|
|
|
|
|
Intrinsic wound
pathologies – auto-immunity versus other theoretical diseases On slides 9 & 32, we asked why the connective tissue and
stromal pathologies are due to autoimmune states as opposed to some other
general class of disease. Throughout
this section we have established the connection between autoimmunity and the
diseases of the general stroma.
However, we have yet to answer the other side of that question: why
are there no common diseases of the fascias, connective tissues, and general
stroma related to metabolic alterations or genetic deficiencies? The full annotated answer is beyond the
scope of this presentation, but a few basics can be explained. The stromal cells, fibroblasts and angiocytes, represent
evolutionary and phylogenetically ancient cells. Multicellular life appeared about 1 billion
years ago, as single celled life learned that there is strength and survival advantage
in cooperative association and the division and specialization of labor. As mentioned already on slide 53, there are
two quintessential constructs needed to permit multicellular association and
function: some system for holding everything together in a stable functional
anatomical form, and some sort of distribution system to permit the
interchange of nutrients, metabolites, and information. In animals, the system that evolved for
holding things together is based on connective proteins, the most abundant of
which is collagen. Collagen structures
and anatomy became increasingly complex as life advanced, but collagen is
present even in the most primitive of multicellular organisms, the Porifera,
the sponges. Evidence of a bulk
transport system – a vascular system – is also seen in some sponges, and it
is permanently established by the Cnidaria, the hydras and jellyfish. Primitive invertebrates do not have a blood
circulatory system. Instead, their gut
has extensions into all parts of the organism to directly deliver food, a
gastrovascular cavity that handles both digestion and distribution. Nonetheless, this is a vascular
distribution network, and our blood circulatory vascular system is a direct
evolutionary descendant of the gastrovascular cavities of the Cnidaria. Only one gene and its product are required
to govern the formation and morphology of this vascular distribution system,
and that gene is VEGF (vascular endothelial growth factor; well, actually 2
genes, VEGF and VEGFR, its receptor).
Genetic sequencing allows us to recognize the specific nucleotide
“spelling” of each gene, and jellyfish and human VEGF and VEGFR are highly
homologous, spelled almost exactly the same.
Also, the observable functions of VEGF on vascular cells and structures
are identical for jellyfish and humans.
As life evolved, many new genes appeared, old ones disappeared, and
many morphed and changed. But, over
eons of multicellular evolution, VEGF and its functions are unchanged. Why? Why has VEGF remained unchanged?
Because multicellular life is wholly contingent on a bulk transport
vascular distribution system. Without
it, complex multicellular life is categorically impossible. Once this core infrastructure element of
life had been written, it needed no revision, because it worked so well. What this means is that for the few
quintessential genes that permit multicellular life, there is little room for
mutation. VEGF is so crucially
essential for life that without it, an embryo unconditionally cannot develop
– period - exclamation. (In some
experiments, VEGF knockout is categorically lethal; in other experiments,
other angiogenic factors can keep a conceptus alive, but with significant
developmental defects.) Whatever VEGF
mutation might occur in a gamete, it cannot be propagated, because a
conceptus simply cannot develop beyond just a few cells (the gastrula
stage). The basic stromal structure of
multicellular life – connective matrix and vascular distribution system – was
worked out from the beginning, 1 billion years ago. The formation and function of these
structures and cells has been thoroughly tested and debugged, meaning they
are essentially error free. These core
infrastructure functions of multicellular life are so consistently conserved
and dependable, so thoroughly robust, that there are no major genetic or
metabolic disorders of the stroma, and consequently none of the mesenchymal
component of wound healing. Because
these cells and structures have extraordinarily few intrinsic disorders, when
wound healing goes bad it reflects some sort of exogenous disorder or damage,
some sort of deprivation or predation affecting these cells and
structures. These extrinsic conditions
include non-specific non-targeted conditions such as trauma, ischemia, toxicities,
and severe metabolic-nutritional inadequacy.
It also includes targeted damage directed against these cells and
structures, and as we have seen here, that means auto-immunity and
lympho-plasmacytic inflammation. |
|
|
|
|
|
This presentation was organized to show first the anatomical and
tissue pathologies that result from the autoimmune diseases, and how they
affect primarily the fibrous and vascular stroma, and thus why they are
called “collagen-vascular diseases” and “connective tissue disorders”. We then looked at why autoimmunity develops
in the first place – due to a sustained inflammatory state that unmasks
immunogenic antigens in the connective and vascular stroma - and thus why it targets the
connective-vascular stroma. We then
demonstrated that autoimmune wounds have altered histological findings,
confirming that wound healing itself is pathological. We will now take the final step of putting
these premises together to show the connection between autoimmunopathy,
connective tissue disorders, and wound healing, and why wound healing is sick
in these disorders. The connections between these states is derived from a pair of
simple syllogisms. At first, it might
seem that a simple mathematical abstraction of these complex
pathophysiologies and clinical syndromes is an injustice to nature and a
disservice to the medical arts and sciences.
But the syllogisms are real, and like most systems in nature, these
concepts can be reduced to simple principles.
The dynamics of complex systems and the physics of self-organization
are discussed in detail in Part 3, after which the simplicity and
mathematical beauty of these syllogisms might be better appreciated. To state the connection between autoimmunity, connective tissue
disorders, and pathological wound healing, we must start with three axiomatic
predicates. (1) All mesenchymal stroma
is composed of 2 cell types and their derivative structures: fibroblasts and angiocytes, blood vessels
and connective matrix. (2)
Wound healing, the proliferative mesenchymal wound module, depends on
2 cell types: fibroblasts and
angiocytes, to remake vessels and matrix.
(3) The target of mesenchymal autoimmune attack
is the collagen-vascular stroma, i.e.
fibroblasts and angiocytes, blood vessels and connective matrix. Assertion 1: The collagen vascular connective tissue
disorders are a disease of wound healing. Basic syllogism : A - Diseases that
affect the collagen-vascular connective stroma ipso facto affect the
mesenchymal wound module. B - The autoimmune
CVD-CTD’s are diseases that affect the collagen-vascular connective stroma. Conclusion: Therefore, the CVD-CTD’s are diseases of
wound healing. Assertion 2: Intrinsic wound chronicity is a collagen
vascular connective tissue disorder. Basic syllogism : A - Autoimmune
diseases that affect the collagen-vascular stroma are the CVD-CTD’s. B - Intrinsic
lymphoid wound chronicity is an autoimmune disorder that affects the
collagen-vascular connective stroma. Conclusion: Therefore, intrinsic wound chronicity is a
CVD-CTD. To reiterate: the
connective tissue disorders are diseases of wound healing; intrinsic wound chronicity is a connective
tissue disorder. Recall what we mean
when talking about “intrinsic wound chronicity”. It is that state where some condition of
sustained acute inflammation has led to stromal auto-immunization and
infiltration with lymphoid cells which continue to sustain wound healing
impairment or inhibition even after the primary disorders have been
alleviated. Why this state becomes
intrinsic, perpetual, and hard to break is the subject of Part 3. Connective tissue disorders and intrinsic
wound failure are essentially the same thing, different facets or avatars of
sensitization or predation against the cells and structures of the stroma. The same tissues that are the targets of
the collagen-vascular diseases are therefore apt to become the casualties of
wound healing, the places where skin ulcers and musculoskeletal ruptures
occur, the places where surgery is likely to have complications, the places
where wound healing is retarded or incompetent. The adverse effects of stromal
auto-immunization and the connective tissue disorders can affect all phases
and states of the wound, because regardless of what phase it is in, it is
still just stroma. This causes one of
the most perverse and pernicious aspects of auto-immunopathic wounds compared
to other diagnoses, the duality of their effects: (1) they have an afferent effect to cause
necrosis and ulceration, and then (2) they have an efferent effect to
continue injuring or inhibiting the wound thus preventing healing. These disorders make the wounds, then they
keep them from healing. These effects
continue even after the wound is nominally healed, leading sometimes to
chronic or intermittent cicatritis, localized panniculitis, and recurrent
scar ulcers (a problem most apt to be seen with lupus and its close
allies). Then, there are the remote or
systemic effects against stroma which are the manifestations of the cvd-ctd. We have made the case that the autoimmune connective tissue
disorders are the true intrinsic diseases of wound healing. For those who have never formally studied
wounds, who only have the cursory awareness of the commonly appreciated
wounds (trauma wounds and the “classic 4”), this may sound surprising, But it should not be. If wounds are made of fibroblasts and
angiocytes, then diseases of fibroblasts and angiocytes are the wound
diseases. If you have not been seeing
these wounds and diseases, it is because you have not been looking, merely
ascribing all wounds to a limited set of commonly known diagnoses. All effective care starts with diagnosis
specific therapies, and that means making the correct diagnosis. Once you start to recognize these wounds,
and start to get the good results that come from specific therapies, the
straightforward validity of the syllogisms above will be evident. |
|
|
|
|
|
Understanding
how auto-immunopathy affects wound healing. Synopsis 1 Not all chronic and pathological (CAP) wounds are in a state of
intrinsic chronicity and self-sustaining auto-immunization. Many have
identifiable active primary pathologies that can be treated and the wounds
healed easily enough. But many chronic wounds are difficult to heal because they have
transitioned into a state of lymphoid autoimmune intrinsic chronicity (LAIC). This state
exists when auto-immunization occurs against mesenchymal stromal cells and
their constructs (angiocytes & fibroblasts, vascular & connective
structures). ==================================== The normal response to incidental injury is the sequence : thrombosis
--> acute inflammation -->
repair (wound module). For incidental
injury in healthy subjects, these events are sequential one-shots without
feedback, retrigger, or other sustentation. These sequential events define the wound in time. The physical anatomy of the wound (strata)
defines the wound in space. Acute
inflammation normally operates early in upper strata. Repair cells normally operate later in
deeper strata. This separation in time and space ensure that there is little
admixture between acute inflammatory cells and stromal repair cells. Without
sustained acute inflammation & population admixture, sensitization (and
then chronic lymphoid inflammation) cannot occur. ==================================== Under various conditions of persistent primary disease or
injury, acute inflammation can become sustained. Causative
primary pathologies include chronic injury, thrombosis, allergy, infection,
or anything that triggers sustained inflammation. Sustained acute inflammation (1) creates matrix degradation
debris and increases the load of normally sequestered endocellular elements. These exposed
chemicals are potential auto-antigens that the immune system is naive to and
would ordinarily never see. Sustained inflammation (2) disorganizes timewise & spatial
wound structure, causing an intermix of cells & strata, inflammatory
& repair. Potential
antigens from ongoing degradation now include those from repair elements -
angiocytes, fibroblasts, and their structures. |
|
|
|
|
|
Understanding
how auto-immunopathy affects wound healing. Synopsis 2 As acute inflammation is sustained, there is increased antigen processing
by macrophages and then the appearance of lymphocytes. Eventually
there is antigenic recognition of items normally hidden from immune cells by
time, space, sequestration, & wound anatomy. Autoimmunity occurs when lymphoid cells in the wound eventually
“see” endocellular or other antigens that they should never have seen. (Auto-immunization
against mesenchymal stroma could not have occurred absent sustained acute
inflammation and population admixture.)
This
pathological state is marked by appearance of a 3rd population - chronic
inflammation - lymphocytes, plasmacytes, & eosinophils. Auto-sensitization includes angiocytes & fibroblasts,
vascular & connective stromal elements, because their antigens were in
the melee. Presence &
persistence of lymphoid cells in the vascular locus attest to the
significance of angioid elements as targets of auto-immunity. ==================================== Once the stroma is auto-sensitized, the 3rd population (chronic
lymphoid cells) has three pairs of pathological effects. (1a)
Chronic inflammation effects to perpetuate acute inflammation. (1b)
Autoimmune predation or inhibition against stromal elements. Chronic
inflammation effects: (2a) Afferent effects to create or
sustain injury & ulceration. (2b) Efferent effects to impair
healing. Autoimmune
effects: (3a) Local effects against the reparative stroma. (3b) Systemic effects against stroma in
general. Chronic inflammation can retrigger or sustain the wound as much
as primary injury or disease does. Afferent
effects: Triggering and sustaining
acute inflammation & thrombosis is what causes necrosis, lysis,
ulceration. Efferent
effects: Disorganizing the cell mix,
strata, & interactive biology of the repair process is what impairs
healing. Auto-immunization against stroma causes many pathological and
clinical sequelae (based on specific antibodies, origins, circumstances). Local effects: Limited
effects presenting as localized vasculitis, panniculitis, etc., or as
impairments of wounds & wound healing. Systemic effects: Global
morbidity recognized clinically as nosological entities, e.g. rheumatoid,
lupus, scleroderma, etc. ==================================== Intrinsic wound chronicity and impaired healing are the local
effects of auto-immunity directed against fibro-vascular stroma. The CVD-CTD’s
are the systemic effects of auto-immunity directed against fibro-vascular
stroma. |
|
|
|
|
|
Understanding
how auto-immunopathy affects wound healing. Synopsis 3 Because their auto-immunopathy is against stromal cells, . . . the
CVD-CTD’s have high incidence of ulceration & impaired wound healing. . . . wounds
with lymphoid autoimmune intrinsic chronicity have a high incidence of
CVD-CTD disease or symptoms. Diseases that affect collagen-vascular connective stroma ipso
facto affect the wound module (stroma in a repair phase after injury). The autoimmune
CVD-CTD’s are diseases directed against stroma. Therefore, the CVD-CTD’s are diseases of
wound healing. Autoimmune diseases that affect the collagen-vascular stroma are
the CVD-CTD. Intrinsic
lymphoid wound chronicity is autoimmunity directed against stroma. Therefore, intrinsic wound chronicity is a
CVD-CTD. ==================================== Wound healing is just a connective tissue event, stroma in a
repair phase after injury. Its impairment
is just another manifestation of connective tissue pathology. The two pathologies, classic connective tissue disorders and
intrinsic wound chronicity are essentially the same disease. They are
autoimmunopathy directed against fibro-vascular stroma, both induced by the
same underlying instigating disorders. They are different manifestations, different syndromic avatars
of the same process, . . . one
acting locally, causing stromal inflammation & ulceration, then
inhibiting stromal repair by damage, inhibition or disorganization. . . . the other
acting systemically, causing arthritis, synovitis, serositis, vasculitis,
panniculitis, myofasciitis, etc. ==================================== In autoimmune wounds, impaired wound healing and chronic
inflammation are conjoined, intermixed with
acute inflammation and variable expression or competence of the reparative
wound module and its elements. The connective tissue disorders and lymphoid wound chronicity
are true intrinsic diseases of wound healing. Inflammation
& immunity are targeted against stroma, meaning ipso facto they are
targeted against the wound healing machinery. |
|
|
|
|
|
Autoimmune & immunopathic diseases do double damage to
wounds. They cause wounds and then they
also impair wound healing. As diseases
of fibroblasts & angiocytes, they impair the intrinsic ability of the
wound module to function. Immunopathic
ulcers are a challenge to treat because wound healing is “broken”. Comprehensive management of the wound and the
underlying disease will prevail, and most of these wounds are curable. The caveat is that, because wound healing
is “broken”, many of them take an unusually long time to heal. Healing rates
and times. In support of this
thesis, one need look only at the healing rates or healing times of various
wound diagnoses. The graph on the
slide shows data from my clinic looking at days until healed stratified by
diagnosis. The rheumatoid and other
autoimmune diagnoses take longer to heal.
This is because wound healing is broken in these disorders. For the other diagnoses, generic wound
healing throughout the body is intact, and wounds heal once local
pathologies, injuries, and inhibitors are removed. For the auto-immune disorders, wound
healing itself is impaired. Another
diagnosis where wound healing is intrinsically impaired is radiation injury,
but the cause is always known, and this is just a small fraction of all
ulcers. In contrast, the autoimmune
chronic and pathological ulcers are a very big group of wounds. Again, if you have not appreciated this, it
is because you have not been looking nor being discriminating and exact about
your diagnoses. The pernicious effect of immunopathy on wound healing is also
demonstrated in the table below. This
is Table 4 from the paper Gottlieb ME,
Furman J: Successful Management and
Surgical Closure of Chronic and Pathological Wounds Using Integra®. Journal of Burns & Surgical Wound Care,
3:2, 2004. (The journal is now
Eplasty, the Open Access Journal of Plastic Surgery, at www.eplasty.com. The paper can also be read at the Arimedica
website.). Integra® collagen-gag
matrix is an artificial skin and regenerative scaffold that has many uses in
reconstructive plastic surgery and chronic wound care. This table shows length of treatment time
for 95 patients with chronic wounds, stratified by diagnosis. Integra® is a two-step process: (1) the matrix is placed on the wound and
allowed to regenerate, then (2) skin grafts are placed on the regenerated
neo-dermis. The first set of data,
“Integra-to-skin grafts”, shows the average regeneration time in weeks, the
time between placing the material then placing the skin grafts. It should be noted that Integra® has an
effect to suppress normal wound healing, and instead turn on an embryonic
model of dermis formation. The length
of this phase, overall average 5.3
weeks, is largely independent of diagnosis, and immunopathy had no effect on
this process. However, skin grafts
were not always completely successful, necessitating additional topical care
or secondary skin grafts. This latter
phase of additional care was a matter of normal wound healing and wound
care. The second set of data shows the
time to full healing, full epidermal restitution. The average time to full healing was
roughly 5 – 6 months for most diagnoses, but it was nearly 10 months for
immunopathic and radiation ulcers, the two diagnoses where the local stromal
cells and wound healing are rendered incompetent and intrinsically
dysfunctional. Table
4. Length of treatment Primary diagnosis Integra
– to – skin grafts (weeks) Integra – to – healed (months) . No.
pts mean std No. pts mean std range Macro-arterial 22 5.3 1.2 16 5.0 2.5 1 - 9 Immunopathic 21 5.4 1.6 12 9.6 5.3 2 - 18 Venous / lymphatic 17 4.6 1.3 11 6.2 3.2 2 - 11 Hypercoagulable 6 5.3 2.0 7 5.8 2.1 4 - 9 Mechanical / anatomical 8 5.0 1.3 6 5.2 1.5 3 - 7 Radiation / malignancy 6 7.4 3.9 4 9.8 4.3 5 - 15 Diabetes 5 4.3 1.1 3 6.5 2.1 5 - 8 Unknown 5 4.1 1.4 4 7.0 0.8 6 - 8 Micro-occlusive 1 6.0 - - - 1 4 - - - - - - Trauma and surgery 2 4.6 1.5 1 3 - - - - - - Granulomatous / infectious 2 4.1 1.6 1 2 - - - - - - Total 95 5.3 2.0 66 7.2 4.3 1 - 19 Wound
chronicity. The long duration, long
healing times, and long treatment times of these ulcers means that they are
“chronic” in the common vernacular meaning of that word, i.e. “a long
time”. However, “chronicity” in a
wound has far greater implications than just the common trivial use of that
term. Remember from slide 21 and
following that these problems have pre-ulcerative, then active-early
ulcerative, then chronic-late features.
During early ulceration, you are apt to see disease specific findings,
such as synovitis or panniculitis or cicatritis. As the acute events wind down and the
chronic ulcer persists, the ulcers develop a wide spectrum of changes which
mark them as chronic. These changes
include gross, histologic, biochemical, and behavioral-dynamical features
which characterize CAP wounds as being distinctly different than acute
healthy wounds. In this presentation,
we have been focused on a specific thesis, and thus the discussion of wound chronicity
has focused on sustained pathological dynamics and the appearance of lymphoid
auto-immunity, i.e. “chronic inflammation”.
Here is a brief synopsis of other aspects of wound chronicity. [1] Gross
findings of chronicity are more or less the same for CAP wounds of any
cause. This is analogous to the end
stage liver or kidney – each organ has a generic final pathological
appearance regardless of a priori causes.
The gross features of chronic non-healing wounds, as opposed to the
appearance and physical signs of a healthy healing wound, are obviously very
familiar to anyone caring for them. If
you are unfamiliar with the subject,
look at the many examples shown on slides 11 – 44. Always keep in mind the diagnostic
significance of the thrombo-infarctive versus inflammatory-lytic patterns of
injury. [2] Biochemical features of chronicity may not be as familiar to many
practitioners, but this subject has garnered major attention from wound
research bioscientists over the past decade.
To date, there are many characterizations of chronic versus acute wound
chemistry. In the past few years we
have started seeing many proteomic and gene chip analyses that let us look
directly at what genes are on or off for chronic versus acute wounds. What has been learned in a short time is
that wound biochemistry and genomics for chronic versus acute are
dramatically different, reflecting two entirely different dynamical
attractors. (The concept of “dynamical
attractors” will be presented in detail in Part 3 of this series.) [3] Histologic features of chronicity were
addressed in the past few slides (54 – 59).
The appearance of chronic inflammation and alterations in the wound
module are easily observed and reflect fundamental differences between
healthy and pathological, and between acute and chronic. [4] Behavioral & dynamical features of
chronicity are self evident – if it is not healing, it is chronic. However, throughout this presentation we
have seen why chronicity develops, because of the effects of sustained
primary disease, injury, and inflammation, then the appearance and abnormal
interactions of lymphoid inflammation.
In Part 3 of this series, we will look at the physics that govern the
dynamical effects of lymphoid inflammation. What is important to realize overall is that wound chronicity is
not just a matter of a “long time”. It
is a global concept about what happens in the impaired wound, the chronic and
pathological CAP wound. Keep in mind
that time is an inherent and crucial concept in wound healing. Wound healing is a time-based process. We can assess many biological states by
their incidental values. Many other biological
activities are perpetual. The wound is
different. It comes, it does it’s
thing over so many days or weeks, and then it recedes. When healthy, the whole process is an
efficient composite one-shot derived from constituent one-shot events. Anything that interferes with its inherent
health can only make the one-shot dynamics be perturbed, meaning that time
delays are introduced, and the process gets longer. At some critical point, the pathological
state becomes self-sustaining, and that is the quintessence of wound
chronicity. Everything else are just
markers of the chronic state. |
|
|
|
|
|
Effects of
wound auto-immunopathy: the arrested
wound module. The next few slides demonstrate the clinical consequences to the
wound of stromal auto-immunopathy and connective tissue disorders. We begin by looking at the arrested wound
module. As we have seen,
auto-immunopathic states disorganize the wound module and break wound
healing. This results in prolonged
healing times compared to other wound diagnoses which are extrinsic to the
wound healing process. What does
broken wound healing look like? That
is, what does it look like when it is REALLY genuinely broken? On this and the next slide, you will see 5
patients in whom wound healing is busted to the point that the mesenchymal
components of the wound module simply do not exist to any meaningful or
recognizable degree. Left top: the thigh in a patient with acute lupus
(see slide 25). The skin necrosis is
more thrombo-infarctive in nature, rather than inflammatory-lytic, and she
had hypercoagulable markers along with the immune markers, meaning that both
pathologies are present. Note the
appearance of the femoral fascia.
Fascia fibers and subcutaneous adipose are still visible weeks after
these wounds occurred, with only the slightest hint of a pink blush to
indicate some abortive angiogenesis. Left bottom: the ankle in a patient with chronic
overlooked rheumatoid (see slides 17 and 25).
This wound had been present several years, and there is essentially
zero wound proliferation – you are looking at native anatomy as though the
tissues had been excised just yesterday.
Right: the dorsal foot in a patient with
rheumatoid (see slide 23). Over an
interval of a few weeks of basic care, areolar fascias and tendon sheaths are
still visible, with no wound proliferation over them. |
|
|
|
|
|
Effects of
wound auto-immunopathy: the arrested
wound module. Here are two more patients with auto-immunopathic diseases and
ulcers in whom wound healing is broken and a wound module has failed to
appear. Left: the buttock in a
woman with rheumatoid, 6 months following some injury or ulcerative
event. While there is a pale pink
blush of angiogenesis, all of the fat lobules of the subcutaneous adipose
maintain their native anatomy, texture, and mechanics as though the wound had
been created just 3 or 4 days ago in a normal person. Right: the leg in a woman with severe polymyositis
following minor household trauma (bumping into a bed frame). We have all seen wounds that look like this,
but they are usually acute. If the
injury had occurred just a week ago, and if care had been neglected, and she
then showed up in the emergency room, nobody would be surprised that an
otherwise normal injury and wound could look this way under those circumstances. However, this wound did have basic hygienic
care, and it occurred 4 months ago.
Wound module events are so impaired that even eschar is not fully
separated yet. |
|
|
|
|
|
Effects of
wound auto-immunopathy: treatment with
steroids. If a wound has become chronic and intrinsically pathological due
to lymphoid auto-immunization, then miscellaneous “wound treatments” may not
have any effect or success. In
principle, nothing will prevail short of arresting the cause of the problem,
the chronic inflammatory and auto-immune state. The next few slides demonstrate chronic
difficult-to-heal wounds that responded promptly and thoroughly to treatment
with steroids or other anti-immune drugs. Shown is a 71 year old man with rheumatoid for 20 years, poorly
treated and uncontrolled. Ulcers have
been present for 2 years behind the medial and lateral malleoli on the left
ankle. His hands and wrists are a
mess. He lives in a perpetual state of
arthralgias and stiffness, and it has become such a way of life for him that
he has forgotten what normal is. The
ulcers have obviously had some sort of basic competent topical care, keeping
them clean, hygienic, and free of gross complications and acute inflammation,
but they have not changed much in 2 years.
When seen in consultation, intralesional triamcinolone was injected
into the ulcers and periwound, and the patient was started on prednisone for
systemic control of disease. These
ulcers are prototypical and pathognomonic of ulceration due to rheumatoid,
lupus, and other classic connective tissue disorders. This story continues on the next slide . .
. |
|
|
|
|
|
Effects of
wound auto-immunopathy: treatment with
steroids. Within 10 days of initial treatment, pain and symptoms were
eliminated from the wounds, and generalized pain and stiffness were almost
fully abated from his joints. His
wrists went from just a few degrees of motion to about 40-50 degrees combined
volar and dorsiflexion. MP and IP
synovitis had subsided dramatically.
Other major joint groups had become more mobile and less painful. The ulcers already showed
improvements. At 3 weeks, the medial
ankle ulcers were nearly healed, and the larger lateral wounds had decreased
in measured area by 35%. At 3 months,
all ulcers were healed and stable, and systemic disease remained well
controlled as the patient was transitioned onto more suitable long term
pharmacological management. The key
message here should be clear. Chronic
inflammation is a potent inhibitor or disruptor of the wound module. Control the inflammation, and wound
dynamics should tend toward normal. |
|
|
|
|
|
Effects of
wound auto-immunopathy: treatment with
steroids. This is a 51 year old woman with a 5 year history of mixed
connective tissue disorder.
Scleroderma-crest signs and symptoms are the most overt, but she has
also been designated as lupus, rheumatoid, and mctd. She has had episodes of large vein
thrombosis in leg and arm, and anticardiolipin antibodies are confirmed. History also includes multiple severe drug
allergies and anti-drug antibodies.
This is another good example of a patient with dual pathologies –
hypercoagulable and auto-immune. The
medial malleolar ulcer resulted from a minor household injury. It has persisted as is, inflamed and
painful for 7 months without response to various treatments. Her auto-immune symptoms are active and
have been for the past year. Steroids were the key to successful treatment. The patient reported good control of
disease when she was on methylprednisolone.
However, she was switched to prednisone about a year ago because of
Cushingoid symptoms, which is when disease symptoms became active. The patient is on warfarin already for the
thrombotic history. Note that the
appearance of the ulcer and periwound is inflammatory-lytic, not
thrombo-infarctive. Based on exam and
history, the immune-inflammatory state is likely to be the predominant
pathology. In addition to basic
hygienic topical care and some light compression, the only treatment was an
adjustment of her steroids back to an effective agent and dose. She was started on methylprednisolone 12 mg
daily. As the pictures demonstrate,
periwound inflammation was settled within one week, wound proliferation was
evident at 2 weeks, and the wound was healed shortly after 1o weeks. All other systemic autoimmunopathy symptoms
improved. This case reiterates the
message that chronic inflammation and autoimmunopathy are potent inhibitors
or disruptors of the wound module.
Control the inflammation and the underlying disease, and wound
dynamics should tend toward normal. |
|
|
|
|
|
Effects of
wound auto-immunopathy: Rx with anti-immune
drugs. This is a 25 year old woman with hypercoagulable and immune
markers (same patient as on slide 45-right).
Hypercoagulopathy was considered to be the primary state. She presented with multifocal vascular
stasis and skin infarcts and ulcers.
The patterns of injury, ulceration, and diffuse skin changes were
predominantly thrombo-infarctive.
Histology showed chronic periarteritis, but no signs of primary
leukocytoclastic arteritis or polyarteritis nodosa. For two years, we treated her with warfarin
anticoagulation. This tended to keep
disease quiet, and the various ulcers and skin changes healed at times. However, disease was never completely quiet,
and she would periodically have recurrent events. After two years, disease activity
accelerated. The addition of steroids
was helpful, but not curative. To the
extent that it did help, prednisone doses crept up, but the patient developed
a variety of hyper-cortisolism side effects.
A switch to auto-immune drugs was tried, but azathioprine and others
were ineffective. However, as soon as
the patient was put on cyclophosphamide, the disease was put to sleep, and
she healed. Steroids were withdrawn, and
the process has stayed quiet. Top left (1): The problem at the beginning of the third
year, when disease and ulceration accelerated and became unresponsive to just
anticoagulants. Note the gross
inflammatory signs of dermatitis and panniculitis. Top
right: (2): A few weeks later, the
scene in image 1 is settled a bit with steroids, but not cured. Bottom
left (3): Three months later, the
patient had an intense resurgence of disease with diffuse focal ulceration,
severe pain, and related thrombotic and inflammatory changes in the skin and
lesions. This is when therapy became
more aggressive. Bottom right (4): Seven
months later, a few months into cyclophosphamide therapy, the legs are
completely healed, all inflammatory changes are gone, all thrombo-infarctive
and vascular stasis changes are gone, and general status is improving
including involution of Cushingoid changes. Regardless of what the original primary pathology was, she
ultimately got better only with a potent antimetabolite that is used to
arrest the proliferation of reticuloendothelial and lymphoid cells. This means that in the end the active
pathology and the impaired wound healing were attributable to the chronic
inflammatory state. You have seen the
problem on slides above, a chronic plasmacytic (lymphoid)
perivasculitis. This was not an acute
or neutrophilic vasculitis as one might ordinarily think about the connective
tissue disorders and the classic arteritides.
This is the chronic
pathological wound in all of its perverse dynamical misbehavior, with chronic
inflammation disrupting the wound module. |
|
|
|
|
|
Effects of
wound auto-immunopathy: Rx with
anti-immune drugs. This is a 35 year old man, previously healthy, who recently
became quadriplegic from trauma. He
soon developed multiple ulcers.
Ischial and other pelvic pressure sores were of obvious cause. However, he concurrently developed
literally thousands of small superficial ulcers (dermis-epidermis only)
covering trunk and extremities. Many
were just a millimeter or a few, but some were of substantial size,
especially those on the chest and abdomen.
Non-specific basic wound care was of little value. When seen in consultation, it was obvious
that the lesions were of immune or allergic origin. Histology showed acute & chronic
inflammation, plasmacytes and eosinophils, and an impaired wound module with
lack of fibroplasia (slide 49). The
nominal diagnosis was a bullous pemphigoid type of atopic reaction which had
progressed to latter phases of lympho-plasmacytic auto-immunization and
autonomy. He was started on prednisone
which successfully resolved many of the smaller lesions, but the larger ones
persisted. The patient soon objected
to the obesity which developed while on steroids, so they were withdrawn, and
the patient was started on azathioprine.
All lesions then healed, and did not recur. Azathioprine was continued for 6 months
after all lesions were healed, and then it was gradually withdrawn. Upper raster: a lesion of the left pectoral area below
the clavicle, Middle raster: lesions of
the mid and lower abdomen. Lower raster: a lesion of the right leg above the
ankle. Left column: The lesions
after some initial non-specific hygiene and wound care, but before initiating
specific therapy. Left center: The lesions 5
months later, after having been on steroids for 2 – 3 months. The leg lesion is improved with some
epithelial growth, but it is unstable and still inherently pathological. The other lesions remain pathological with
no net change. (Many of the smaller
lesions on his body did heal in this interval.) This is when azathioprine was started. Right
center: 7 months into treatment, 2
months after starting azathioprine.
The leg lesion is fully epithelialized and stable. The chest and abdomen lesions are healthier
looking and actively healing. Right column: during the subsequent several months, all
lesions healed and have remained that way. |
|
|
|
|
|
We have developed a thesis about stromal auto-immunization and
its detrimental effects on the wound and wound healing. Stromal
auto-immunization occurs from chronic conditions of hypercoagulability,
allergy, repetitive injury, and similar conditions which sustain acute
inflammation. Once auto-immunization
and chronic plasma-lymphocytic inflammation ensue, the pathological state
becomes self-sustaining. Patients
might first develop a generalized stromal auto-immunization, a classic
connective tissue disorder which can also have subsequent wound healing
impairments. Alternately, chronic
sustained wound problems might lead first to local wound auto-sensitization,
which then might or might not lead to some systemic immunopathy
symptoms. Key to the commonality of the connective tissue disorders and
chronic auto-immune intrinsic wound failure is that both are disorders of the
generic stroma, the body’s basic support framework of vessels and connective
proteins made by angiocytes and fibroblasts.
When inflammation & immunity are targeted against stroma, they are
ipso facto targeted against the wound healing machinery. Simply put, (1) the CVD-CTD are diseases of
wound healing, and (2) intrinsic wound chronicity is a CVD-CTD. Wound healing is a primordial system in
multicellular life which has few metabolic, genetic, or degenerative
defects. It is stromal
auto-immunization, the connective tissue disorders, and lymphoid wound
chronicity which are the true intrinsic diseases of wound healing. With those basic points of the thesis in
mind, here for your consideration are some corollaries and calls to action: Defining wound
chronicity as a distinctive entity.
The concept of the chronic wound as a distinct pathological and
clinical entity, different than acute and healthy wounds, is something that
must be clearly enunciated in textbooks, curricula, and other tools of wound
education. (The fact that formal
education about wounds barely exists at all is another concern.) Differences between acute and chronic
wounds are being studied by modern day laboratory researchers largely by
looking at variances in biochemistry and genomics. Such investigations are important, but the
result is or will be just a giant chart of changes, thousands of individual
items that go up or down in the chronic wound. Such a list of particulars offers no insight
as to how the many items inter-operate in the altered multi-control system of
the chronic wound, nor which are cause, which are effect, and which are
passive inconsequential changes. Like
anything else in medicine, understanding how to treat and get better results
begins with a more thorough understanding of wound anatomy, histology,
pathophysiology, cell and tissue biophysics, and clinical presentation. These are the subjects that truly define
the chronic and CAP wound as a nosological entity, and they need more robust
development and teaching. Defining wound
chronicity as a CVD-CTD. Even if
the idea of the wound as a connective tissue disorder seems odd, it will not
as you truly study wound pathology, make accurate diagnoses, and pick the
most relevant treatments. Keep in mind
the well known effects of the defined connective tissue disorders to make
wounds and impair their healing. Keep
in mind also the central importance of the generic stroma – angiocytes,
fibroblasts, vessels, and connective matrix – the common thread between the
body structure, wound healing (the stroma repairing itself after injury), and
the collagen-vascular diseases (the disorders that affect the stroma). As part of defining the chronic wound as a
distinctive entity, there is a need to identify and clarify the clinical
profiles and laboratory serologies that will allow the chronic wound to be
approached, diagnosed, and monitored the same as we do the other connective
tissue disorders. For wounds which
have become “intrinsified” by chronic lympho-plasmacytic inflammation,
anti-inflammatory and anti-immune drugs have a crucial role in treatment, and
we collectively need to study and define the best therapeutics. We need to match specific clinical and
laboratory profiles of the chronic intrinsified wound to specific anti-immune
drugs and treatment strategies for the best results healing those wounds. Defining
CVD-CTD as an intrinsic disease of wound healing. The subject of wounds, for all the interest
and enthusiasm it has generated in the past 10 years, remains only
quasi-professional and quasi-informed in its approach to wound science and
the clinical arts. Unlike most
traditional medical specialties which demand knowledge, precision, and
accuracy in diagnosis and treatment, “Woundology” seems willing to settle for
much less. The lack of a robust
physician level curriculum, the lack of formal medical education and training,
and an insufficient number of those professorially ranked on this subject keep
many practices limited to the simplistic diagnostic misappropriation of all
wounds to the “classic 4” of arterial-venous-diabetic-pressure. There is a wide spectrum of diseases and
disorders which cause chronic and pathological ulcers, and until physicians can start to make comprehensive
and correct diagnoses, “wound care” will continue to be much less, and much
less effective than it should and could be.
Among the various categories of disease which cause problem wounds,
the auto-immunopathies and connective tissue disorders are not only one of
the largest but also one of the most relevant to everyday practice and one of
the most challenging to get good results.
The thesis presented here goes beyond the mere appreciation of CVD-CTD
as a cause of chronic ulcers to a deeper understanding of the commonality of
the anti-stromal diseases and the essential equivalence of CVD-CTD with
certain impaired wound states. It is
time to break the anachronistic parochialism of legacy wound “practice” and
start to correct the errors of wound misdiagnosis and consequent mistreatment. The
interconnections between hypercoagulability & auto-immunity. We have seen here how stromal auto-immunization
can occur from a number of primary disorders which create sustained injury,
allergy, thrombosis, and acute inflammation.
The association is particularly demonstrable for the hypercoagulable
disorders, and many chronic wounds are due to a connective tissue disorder or
a hypercoagulable disorder or both.
The connection between hypercoagulability and auto-immunopathy is real
and it is causative. Hypercoagulability
causes autoimmune disorders such as rheumatoid and lupus. It is not the exclusive cause, and other
chronic disorders (injury, inflammation, allergy, infection) can have the
same effect. Nonetheless, this is a
major association of pathological states and diseases, causes and effects
which warrants substantial scientific investigation and revision of
conventional medical thinking. Auto-immune
intrinsic chronicity and the physics of wound failure. Simply put, intrinsic wound pathology and
chronicity is a dynamical disorder of complex populations caused by
auto-immunopathic disruption of the wound module. The “dynamical” and “complex populations”
aspects of this will be explained in Part 3 of this series. For this, Part 2, the conventional medical
part that looks at traditional pathology, the message is about the
commonality of the anti-stromal diseases.
This commonality implies the equivalence of CVD-CTD and those chronic
wounds rendered intrinsically pathological by the effects of stromal
auto-immunization and lympho-plasmacytic chronic inflammation. Here are two additional related items not on the slide: Research needs. While this presentation is not discussing
therapies and management, this slide is a good moment to mention a serious
clinical research need in wound practice, the need to tailor therapies to
specific flavors of immunopathy. Just
as rheumatology research has identified which of many anti-inflammatory and
anti-immune therapies are best suited for specific nosological diagnoses, so
too we in wounds have a need to match specific immunopathies, dermatoses, and
wound profiles to the most effective anti-immune drugs for healing those
wounds. Localized
versus systemic auto-immunity.
This is also a good place to mention auto-immune wounds versus
generalized auto-immune states. If a
patient becomes sensitized to components of the connective stroma, then a
global connective tissue disorder and inflammatory state could occur. That is certainly the case with acute
rheumatic fever, acute lupus, acute and chronic rheumatoid arthritis,
etc. For patients with chronic wounds,
some do have active generalized inflammatory states or classic
rheumatological diagnoses. However,
some seem to have clinical effects just on wounds and wound healing. It is likely that for many of them, they
are sensitized or immunized to occult antigens, or else they breed unique
lymphocyte and plasma cell “hives”, either event occurring only locally and
incidentally in the wound itself. These
would be antigens unmasked or lymphocytes bred during conditions of acute
injury, inflammation, thrombosis, allergy, or wound healing, with activities
directed against the debris of localized thrombosis, inflammation, vascular
and matrix degradation, and the proliferation and degradation of angiocytes
and fibroblasts. For the
research-minded among the wound healing brotherhood, there is a lot of work
to be done to identify the details of all of this. |
|
|
|
|
|
This slide is a reminder that this is a three part presentation
that looks at wound pathology from the point of view of its applicable
physics, elucidating the intrinsic dysfunctions of the wound as a result of
dysdynamia, especially when stromal auto-immunization has occurred due to
prolonged population admixture in a repetitively injured wound. Part 1 – The Wound as a System and a Controlled Machine Part 2 – Auto-Immunopathy and the Intrinsic Disease of Wound
Healing Part 3 – Chronicity and the Physics of
Wound Failure These presentations and supplementary
materials are all available at www.arimedica.com . |
|
|
|
|
|
Summary of autoimmunopathy
and wound chronicity. The connective tissue disorders and collagen vascular diseases
(cvd-ctd) and pathological wound chronicity are essentially the same disease,
and they are the true intrinsic diseases of wound healing. These are the disorders which target
stromal cells & structures – fibroblasts and angiocytes, vessels and
connective tissues – which are also the principal agents of wound healing. This pathology arises when sustained primary injury or disease
causes sustained acute inflammation, which leads to wound disorganization and
sensitization against auto-antigens. Auto-sensitization
will occur against whatever is in the sustained wound, with stromal elements
consistently at risk. Stromal
auto-sensitization and lymphocytic wound chronicity (plus sustained primary
disease and inflammation) cause ulceration and disrupt healing. This and the cvd-ctd’s are a major category
of chronic wounds and wound pathology.
Since these disorders affect the primary agents of wound healing,
wound healing is more impaired than for other causes of chronic wounds which
are extrinsic to the primary process. |
|
|
|
|
|
End |
|
|
|
|
|
Abstract (as submitted in advance of the meeting) The Physics and Pathology of
Wounds. Part 2. Auto-Immunopathy and the Intrinsic
Disease of Wound Healing. Marc E. Gottlieb, MD, FACS Phoenix, AZ Many chronic wounds result from
disorders extrinsic to the healing process, e.g. pressure or arterial
disease. What then are the intrinsic diseases of wound healing? Compare the
wound to other organs. The quintessence of heart failure is that it is an
inadequate pump, for lung failure it cannot exchange gases. But the wound is
neither pump and pipes, nor bellows and diffusion membrane, nor is it like
any organ with macro-anatomical structure. It is a transient collection of
mutually interacting self-organizing cells.
Stromal angiocytes and fibroblasts (wound cells) have remarkably few
inherent metabolic or genetic faults. Dysfunction of the aggregate population
is almost always the result of deprivation or predation. Adverse states can
be caused by (1) non-targeted exogenous conditions such as arterial ischemia
or repetitive trauma, and (2) targeted damage directed against these cells
and their structures. As will be presented here, predation against the wound
module is due to a state of auto-immunopathy in which lymphoid cells are
sensitized to wound components. Not only does this occur with classic
connective tissue disorders and other well-recognized auto-immunopathies, but
it happens when a wound becomes intrinsically chronic and pathological. Hypercoagulability and other conditions of
persistent thrombosis and acute inflammation are the underlying states that
induce the auto-immunization. Simply put, intrinsic wound pathology and
chronicity is a dynamical disorder of complex populations caused by
auto-immunopathic disruption of the wound module. In these chronic non-healing wound
samples, the vascular locus is infiltrated with immune cells (left, plasma
cells; middle, plasma cells and eosinophils; right, lymphocytes.) On the left, plasma cells are mixed with
the migratory angiocytes (spindles) that are trying to assemble the
wound. At middle and right, chronic
thrombosis due to a primary hypercoagulable disorder is not only present, it
is the root cause of this entire state. |
|
|
|
|
|
THE PHYSICS AND
PATHOLOGY OF WOUNDS. PART 2. AUTO-IMMUNOPATHY
AND THE INTRINSIC
DISEASE OF WOUND HEALING. Original presentation February 22 - 26, 2010, Maui, at the The John A. Boswick, M.D. Burn and Wound Care Symposium 2010 The presentation and related materials are accessible at: arimedica.com Content may be used for non-commercial educational purposes. Content may not be published or used for commercial purposes
without prior license or permission. Contact information is on the slide. Copyright © 2009, Marc E. Gottlieb, MD Revision 01a, February
22, 2010 |
|
|
|
|
|
These presentations and supplementary
materials are all available at www.arimedica.com. The Physics and Pathology of
Wounds. Part 1. The Wound as a System and a Controlled
Machine. Marc E. Gottlieb, MD, FACS Phoenix, AZ The wound is a transient organ of
inter-operating cells, triggered into being by injury and inflammation, then
extinguishing as it completes its repair of injured stroma. It is a system.
Conventional bioscience tends to characterize properties and interactions of
individual or one-versus-another elements within a system, but physics is
required to understand the integrated timewise behavior of whole systems.
Intrinsic wound pathology and chronicity, and wound failure and therapeutics
are easily explained when wounds are seen as a non-linear System (rather than
as a collection of dual-element linear interactions). For normal wound
physiology and for the pathophysiology of altered and failing wounds, the
governing principles are the physics of complex systems: non-linear N-element
dynamics, control science, population logistics, and self-organizing
automata. Understanding wound physics begins by
characterizing normal wound physiology. The wound is a closed-loop
reference-driven non-linear multicontrol system. Sick and altered wounds have
layers of added complexity, but the quintessential intrinsic machinery of
wound healing – the Wound Module of post-inflammatory wound repair –
functions as just a single control loop. When tissues are injured, the Main
Control Loop of physiological wound repair will drive cells to reorganize
back to a repaired stroma. The wound control system is composed
of these elements: The system state is the open wound and its conditions. It
is compared (?) to a reference, normal epithelialized tissue. Variances
generate an error signal in the form of inflammation. This activates
macrophages which are the system controller. They in turn generate a control
signal in the form of cytokines. The controlled load is the group of local
responder cells. Their output are the elements of histogenesis, which modify
the state of the system, which then feeds back to the loop at the summing
point. Any discussion or research of the collective behavior of a wound must
acknowledge this basic control system. |
|
|
|
|
|
These presentations and supplementary
materials are all available at www.arimedica.com. The Physics and
Pathology of Wounds. Part 2. Auto-Immunopathy and the
Intrinsic Disease of Wound Healing. Marc E. Gottlieb, MD,
FACS Phoenix, AZ Many chronic wounds
result from disorders extrinsic to the healing process, e.g. pressure or
arterial disease. What then are the intrinsic diseases of wound healing?
Compare the wound to other organs. The quintessence of heart failure is that
it is an inadequate pump, for lung failure it cannot exchange gases. But the
wound is neither pump and pipes, nor bellows and diffusion membrane, nor is
it like any organ with macro-anatomical structure. It is a transient
collection of mutually interacting self-organizing cells. Stromal angiocytes and fibroblasts (wound
cells) have remarkably few inherent metabolic or genetic faults. Dysfunction
of the aggregate population is almost always the result of deprivation or
predation. Adverse states can be caused by (1) non-targeted exogenous
conditions such as arterial ischemia or repetitive trauma, and (2) targeted
damage directed against these cells and their structures. As will be
presented here, predation against the wound module is due to a state of
auto-immunopathy in which lymphoid cells are sensitized to wound components.
Not only does this occur with classic connective tissue disorders and other
well-recognized auto-immunopathies, but it happens when a wound becomes
intrinsically chronic and pathological.
Hypercoagulability and other conditions of persistent thrombosis and
acute inflammation are the underlying states that induce the
auto-immunization. Simply put, intrinsic wound pathology and chronicity is a
dynamical disorder of complex populations caused by auto-immunopathic
disruption of the wound module. In these chronic non-healing
wound samples, the vascular locus is infiltrated with immune cells (left,
plasma cells; middle, plasma cells and eosinophils; right, lymphocytes.) On the left, plasma cells are mixed with
the migratory angiocytes (spindles) that are trying to assemble the
wound. At middle and right, chronic
thrombosis due to a primary hypercoagulable disorder is not only present, it
is the root cause of this entire state. |
|
|
|
|
|
These presentations and supplementary
materials are all available at www.arimedica.com. The Physics and Pathology of
Wounds. Part 3. Chronicity and the Physics of Wound
Failure. Marc E. Gottlieb, MD, FACS Phoenix, AZ The wound module is a transient set of
interacting cells which collectively restore injured tissue to normality, a
fibrous stroma of angiocytes and fibroblasts. Its healthy aggregate behavior
is a well behaved machine, governed by the physics of control systems. A sick
system can result from various extrinsic perturbations, but the core
mechanism of self-sustaining persistent dysfunction, the true intrinsic
disease of wound healing is chronicity itself, the paramount cause being
wound module autoimmunization. This state is disruptive but not fully toxic
or lethal, thus immunopathic wounds have complex behaviors, at times
better-worse-stable-variable, often looking healthy, but always frustrating
as they refuse to cross the finish line. How does one explain such variable
behavior and the differences between normal and chronic-and-pathological
(cap) wounds? Simply stated, intrinsic wound
pathology and chronicity is a dynamical disorder of complex populations. The
physics governing complex behaviors in complex systems is non-linear dynamics
(nld). In addition to control, three aspects of NLD are especially important
to wound pathology. (1) Population logistics. Healthy healing is a sequence
of one-shot self-completing linear events: primary injury & thrombosis
–then– acute inflammation –then– wound module. Pathology creates abnormal
population dependencies (nutrition, starvation, predation, cultivation) and a
new population, chronic inflammation. Non-linear perpetual complexity arises
in the logistics of injury & thrombosis –vs– acute inflammation –vs–
wound module –vs– chronic inflammation –vs– injury & thrombosis. (2) Cellular
automata & self-organization. The “cellular” agents of the wound module
(real biological cells in this case) have a small set of deterministic rules
of interaction with each other. When
allowed to function properly, stromal rebuilding is automatic and
correct. Under pathological
conditions, self-organization, i.e. wound healing is disrupted. (3) Chaos & N-body dynamics. The net
effect is that the wound, a set of several interacting cell populations, has
3 attractors (basins, dynamically stable states or behaviors): convergence
(healing), divergence (ulcerating), and self-sustained chaotic orbits
(chronicity). Basic methods to demonstrate
non-linear dynamics: left, the logistical map of competing populations;
middle, diffusion-limited-aggregation, an example of self-organizing
automata; right, attractors and chaos in the Mandelbrot set of complex-plane
iteration. While seemingly abstract,
these structures are directly correlated with wound events. |
|
|
|
|
|
|
|
|
|
|